En lesiones únicas el tratamiento es curetaje e injerto óseo cuando sea necesario.
Definición: Lesión no neoplásica de etiología desconocida, caracterizada por una intensa proliferación de elementos reticulohistiocíticos con un número variable de eosinófilos, neutrófilos, linfocitos, células plasmáticas y células gigantes multinucleadas. Frecuentes áreas de necrosis, así como presencia de células grasas, especialmente en lesiones antiguas y múltiples.
Incidencia: La reticuloendoteliosis presenta varias formas de afectación, pero se divide principalmente en tres formas básicas: Granuloma eosinofílico (75%), Hand-Schuller-Christian (15%) y Letterer-Siwe (10%).
Granuloma eosinofílico: 5 a 20 años
Hand-Schuller-Christian: 3 a 5 años
Letterer-Siwe: 1 a 3 años
Etiología: La reticuloendoteliosis no tiene una etiología conocida, sin embargo algunos autores la relacionan con una probable causa viral o inmunológica, debido a la presencia de un fenómeno inflamatorio con formación de un proceso granulomatoso hiperplásico, muchas veces similar a procesos neoplásicos.
Manifestaciones Clínicas: La historia natural de la evolución de esta enfermedad dependerá de una de las tres formas en que se presente.
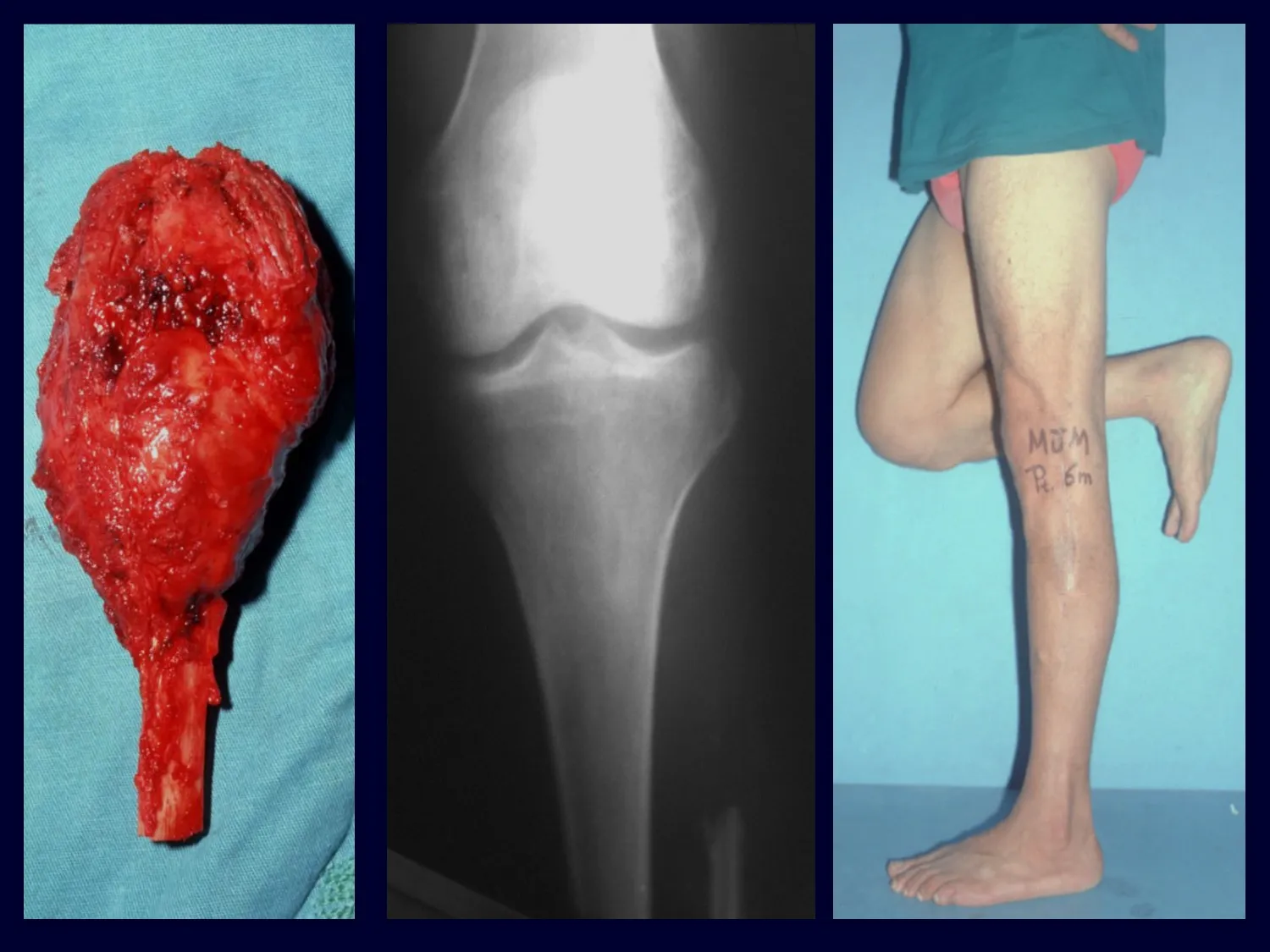
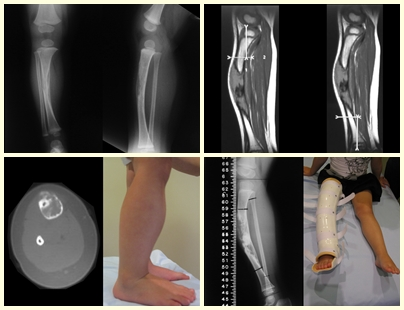
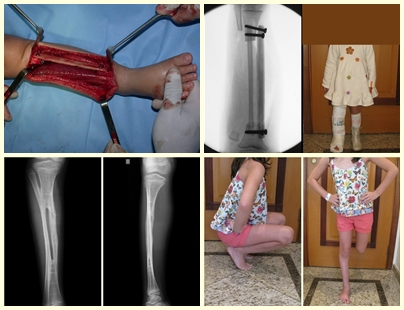
– Granuloma eosinofílico: la mayoría de las veces se presenta como una lesión única, afectando preferentemente a la región diafisaria y metafisaria de los huesos largos, y más raramente también vemos casos con afectación múltiple, que puede ser simultánea o consecutiva, comenzando en la adolescencia y arrastrando. hasta la edad adulta joven. Las lesiones únicas suelen acabar resolviendo espontáneamente con el tiempo, que va de meses a años, y rara vez son incapacitantes o conducen a una fractura patológica.
– Hand-Schuller-Christian: normalmente cursa con múltiples lesiones, que son más difíciles de tratar y evolucionan de forma más incapacitante que el Granuloma Eosinofílico. Frecuentemente presentan afectación secundaria de otros tejidos, progresando frecuentemente a Diabetes insípida (afectación de la glándula parapituitaria), exoftalmos por afectación orbitaria y afectación del hígado y del bazo.
– Letterer-Siwe: los hallazgos clínicos más frecuentes son fiebre, otitis media y antecedentes frecuentes de infecciones bacterianas, y en algunos casos hay anemia, hepatoesplenomegalia, sangrado sin causa aparente, linfadenopatías y lesiones óseas diseminadas. La evolución suele ser fatal debido a la afectación sistémica.
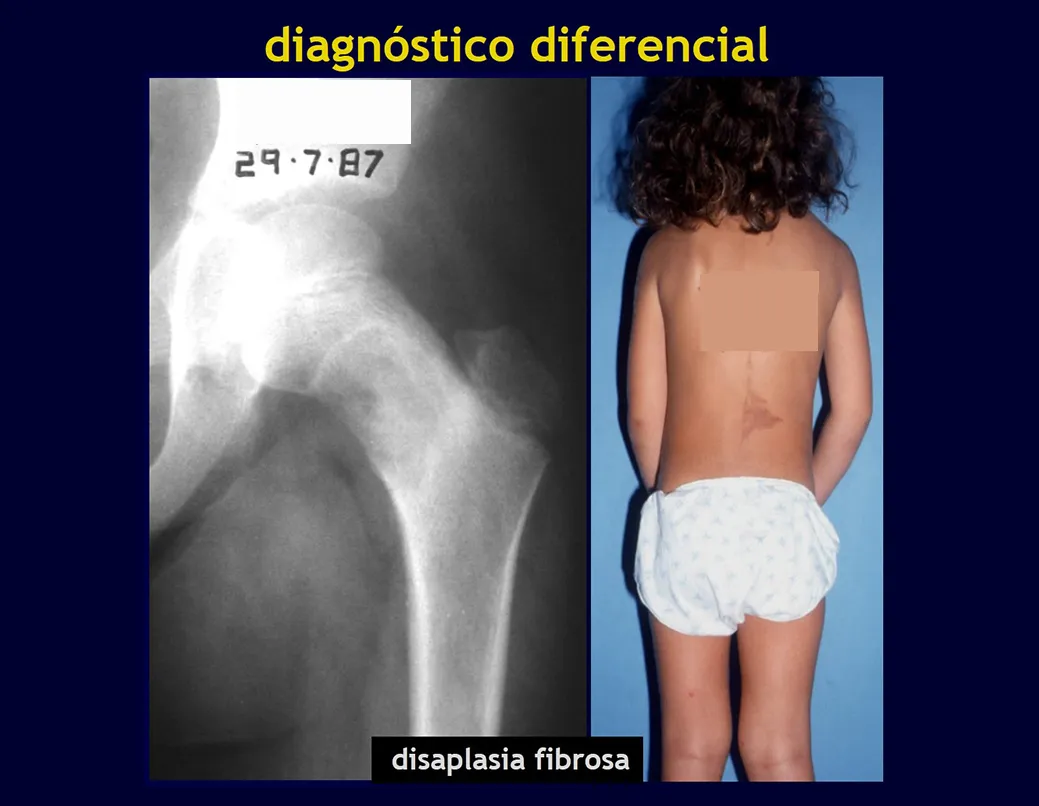
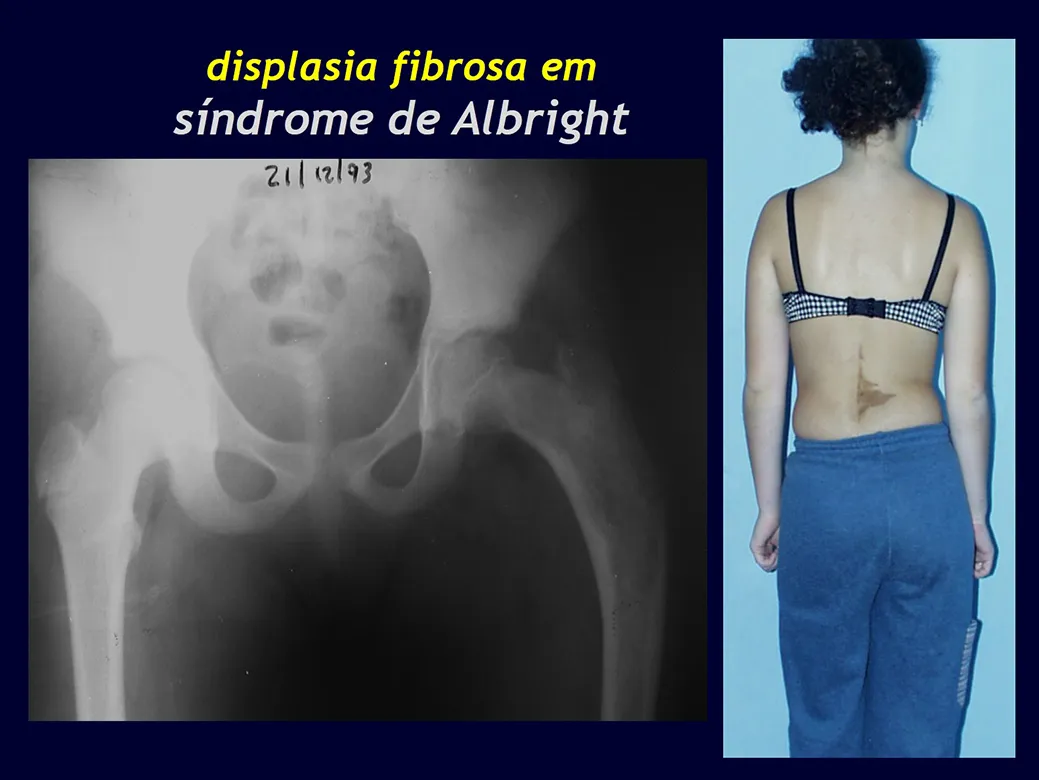
Aspectos radiográficos: Las lesiones tienen un aspecto radiotransparente, con forma redondeada y ovoide, con bordes bien definidos y bien definidos, y en muchas ocasiones pueden verse trabéculas en su interior. Con frecuencia afectan la región diafisaria de los huesos largos y con menos frecuencia la región metafisaria, provocando erosión cortical y ligera expansión cortical. Es posible visualizar un pequeño levantamiento perióstico con una reacción de “piel de cebolla” similar a la del Sarcoma de Ewing y la osteomielitis.
Cuando la afectación es en la columna, rara vez deriva en deterioro neurológico, aunque se produce un colapso de la vértebra, presentándose un aplanamiento y conocido como “vértebra plana de Calvé”.
En casos más graves, como el síndrome de Hand-Schüller-Christian y el síndrome de Letterer-Siwe, se observan lesiones radiotransparentes diseminadas en la bóveda craneal.
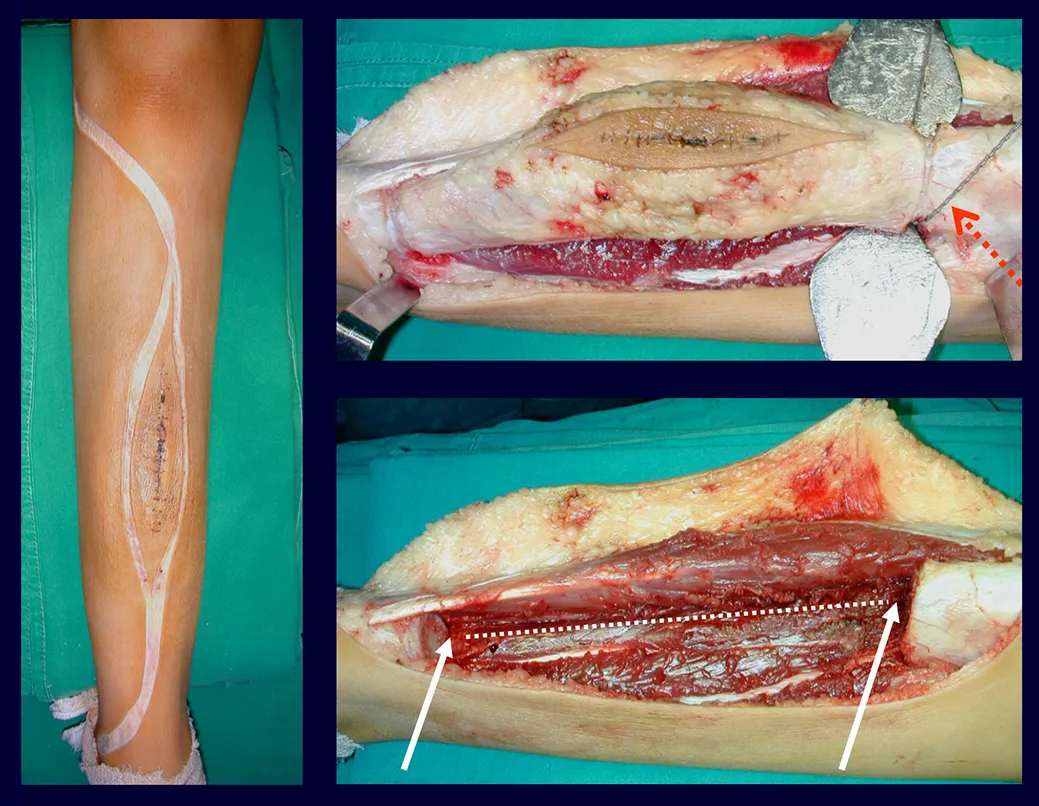
Tratamiento y Pronóstico : El tratamiento y pronóstico de la enfermedad dependen directamente del grado de afectación y manifestaciones clínicas. En lesiones únicas el tratamiento de elección es el legrado y en defectos grandes el relleno con hueso esponjoso. En algunos casos donde no hay deterioro funcional o estético, se puede realizar la resección del hueso comprometido, como las costillas, la clavícula y la parte superior del peroné. En casos de afectación múltiple y sistémica, parte del tratamiento se realiza con el uso de fármacos quimioterapéuticos y terapia con corticosteroides.
1- Haz clic para ver más: http://bit.ly/granuloma_eosinófilo-por
2- Caso de granuloma eosinófilo poliostótico : http://bit.ly/Granuloma_Eosinófilo_do-Rádio
Defecto fibroso cortical / Fibroma no osificante
El defecto fibroso cortical es una lesión ósea benigna, no neoplásica, de causa desconocida, que se caracteriza por una proliferación fibrosa en una pequeña zona del hueso cortical. El fibroma no osificante es el mismo proceso, con un tamaño mayor.
El defecto fibroso cortical generalmente no presenta ningún síntoma o signo clínico. En la gran mayoría de los casos se diagnostica mediante un examen radiológico realizado por algún motivo. Cuando adquiere las características de un fibroma no osificante, puede manifestarse como un dolor leve, una protrusión perceptible por el paciente o, con menos frecuencia, una fractura.