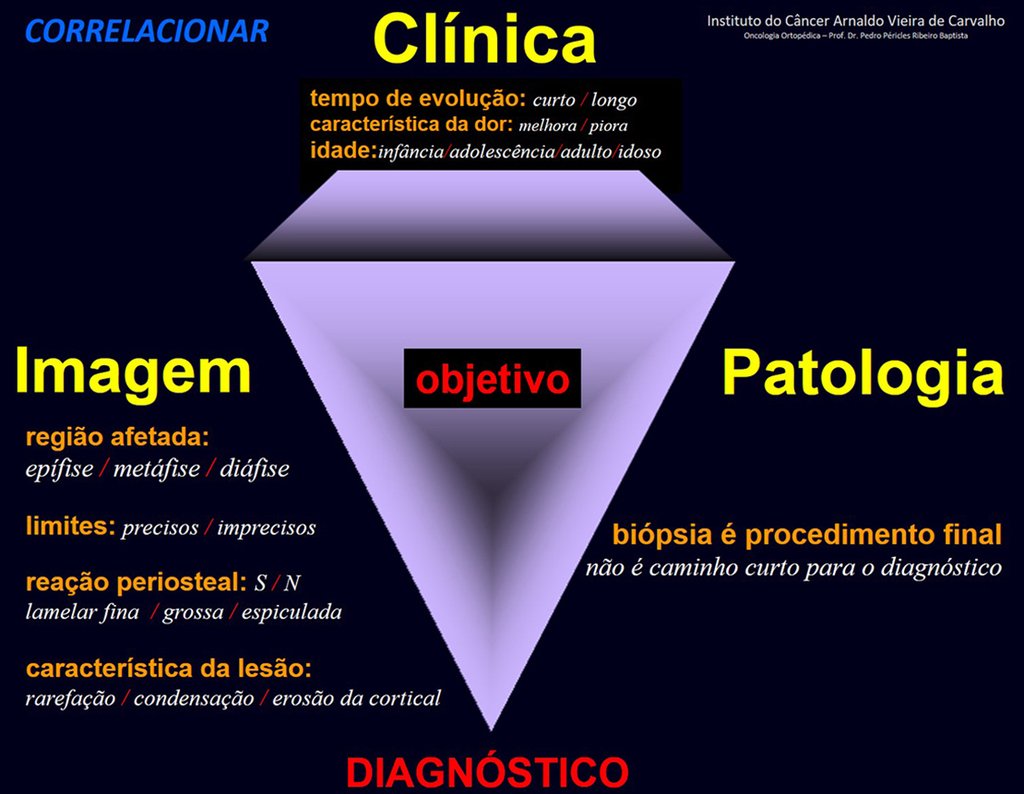
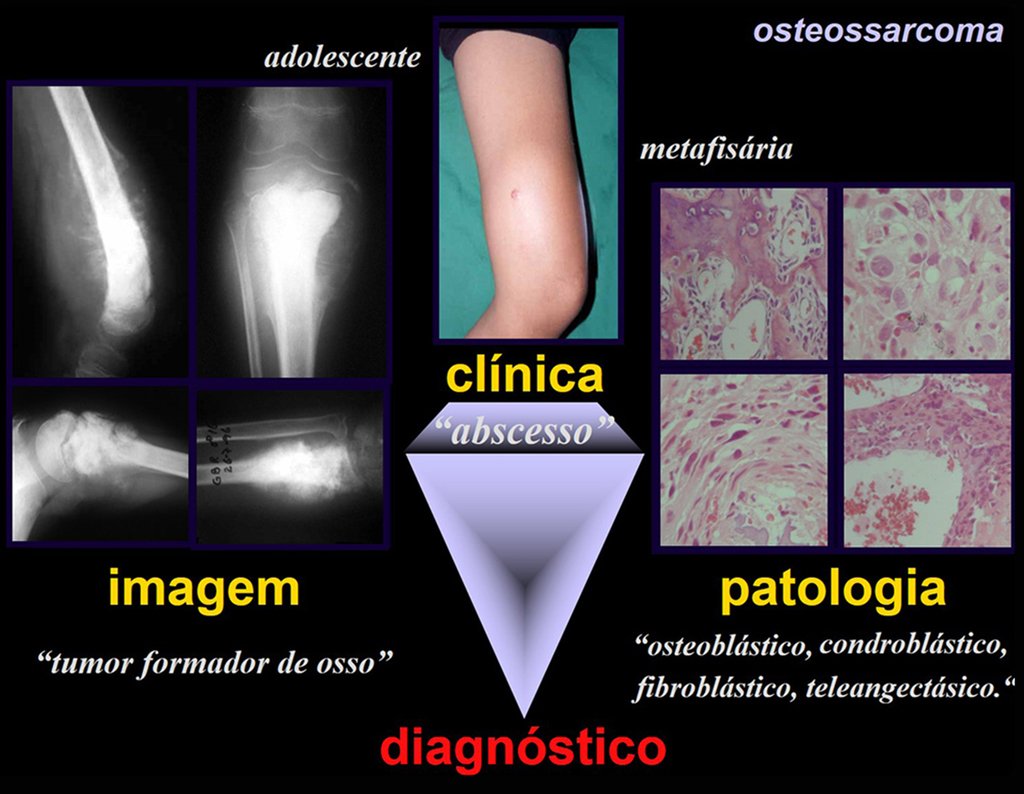
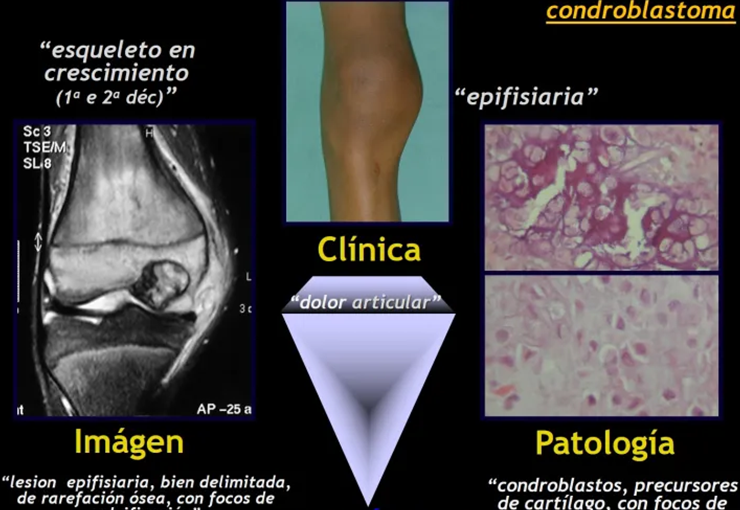
Tumor de células gigantes.
El tumor gigantocelular es una neoplasia de naturaleza mesenquimal, caracterizada por la proliferación de células gigantes multinucleadas –gigantecitos– que se asemejan a los osteoclastos, en medio de un estroma de células mononucleadas.
Debido a este aspecto histológico presente en varios otros procesos, el tumor gigantocelular podría confundirse, requiriendo muchas veces el análisis del aspecto clínico y radiológico para confirmar su diagnóstico.
El tumor de células gigantes también se conoce con las siglas TGC, con los nombres de tumor de células gigantes y osteoclastoma.
La principal manifestación es el dolor local intermitente, acompañado o no de aumento de volumen en la región afectada y limitación del movimiento en la articulación vecina. La duración de la historia, en promedio de 6 a 12 meses, varía de un caso a otro y depende del hueso afectado.
Algunos pacientes buscan tratamiento por dolor, otros por la percepción del tumor o de una fractura patológica. No es raro hacer referencia a un traumatismo más o menos intenso como inicio de la historia clínica.
Como el tumor suele ser epifisario, es frecuente la afectación clínica de la articulación vecina, con impotencia funcional progresiva.
Puede producirse derrame articular y síntomas clínicos que simulan procesos meniscales o artríticos.
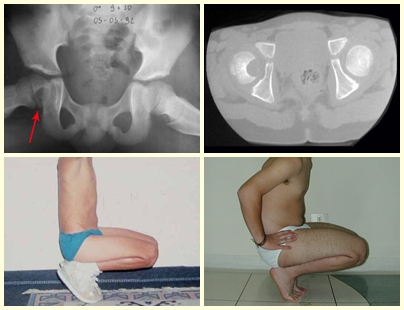
En los tumores localizados en la columna y el sacro, además del dolor y el aumento de volumen, pueden presentarse manifestaciones neurológicas. El volumen es a veces enorme, predominando sobre los demás síntomas.
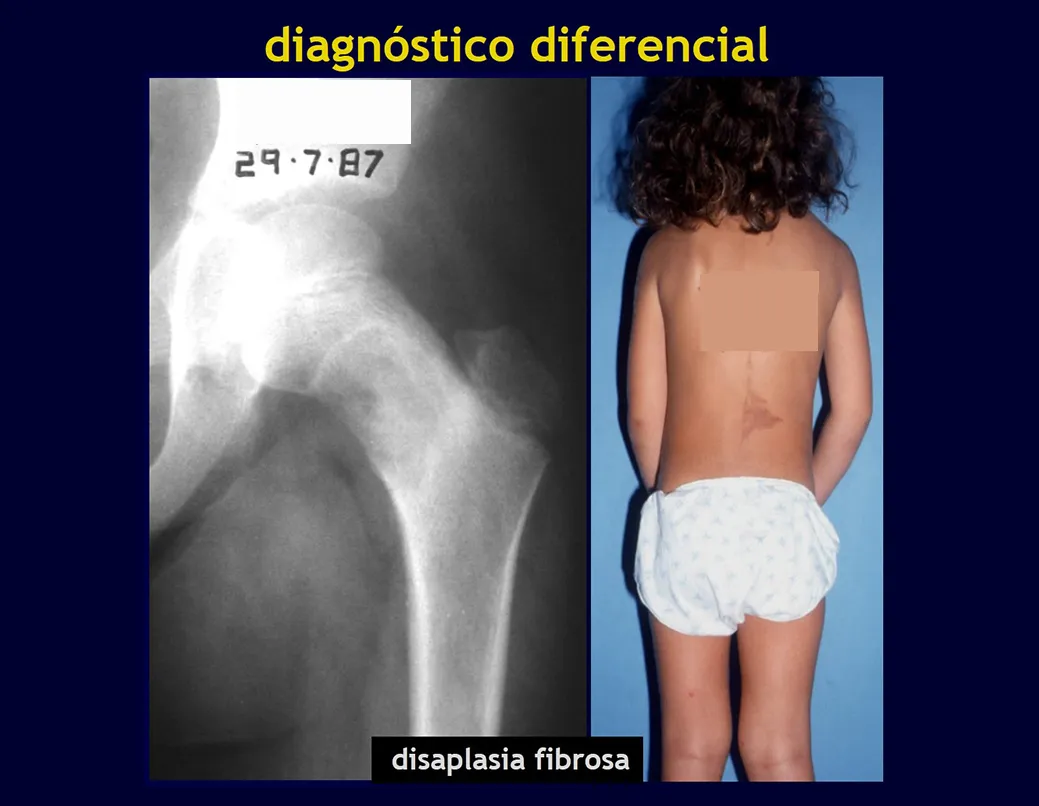
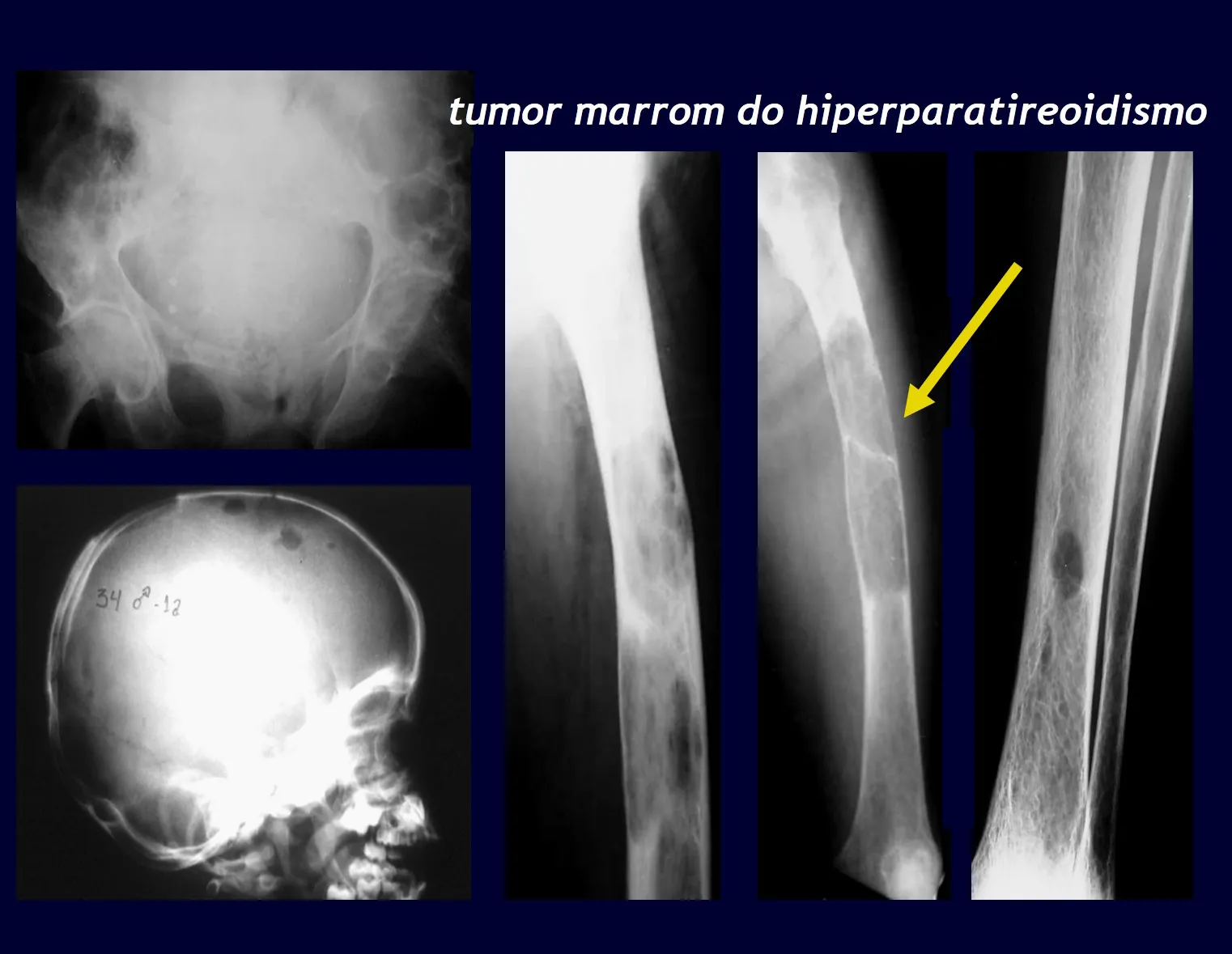
El tumor gigantocelular afecta generalmente a un solo hueso. Cuando existen lesiones en varios huesos se debe comprobar la posibilidad de que se trate de un “tumor marrón de hiperparatiroidismo”, cuyas lesiones tienen aspectos radiológicos similares, pero son múltiples y el paciente presenta alteraciones en el calcio y el fósforo.
El TCG se conoce clásicamente como un tumor de la epífisis de los huesos largos, más frecuente en la región de la rodilla, es decir, en la epífisis distal del fémur y epífisis proximal de la tibia y, luego, en orden de frecuencia, en la región proximal. del húmero y región distal del radio. Es raro en el esqueleto axial y cuando ocurre predomina en el sacro.
Ocurre en la tercera y cuarta década, afectando a ambos sexos por igual.
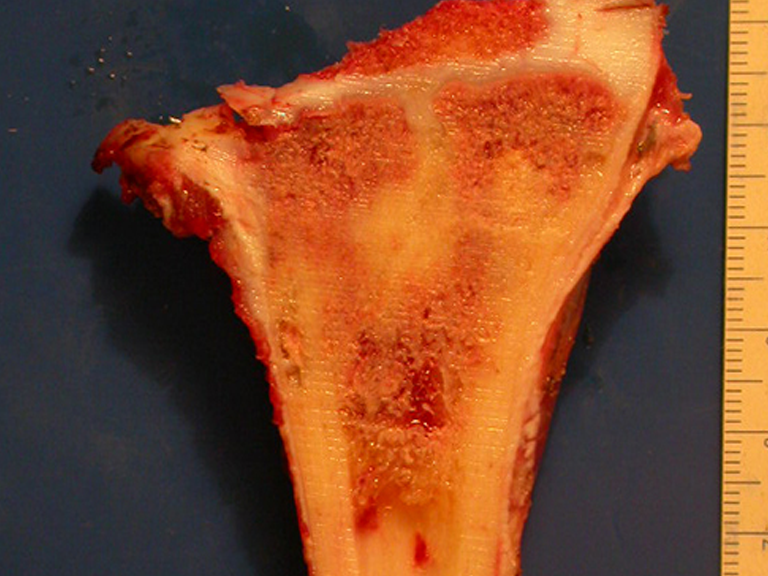
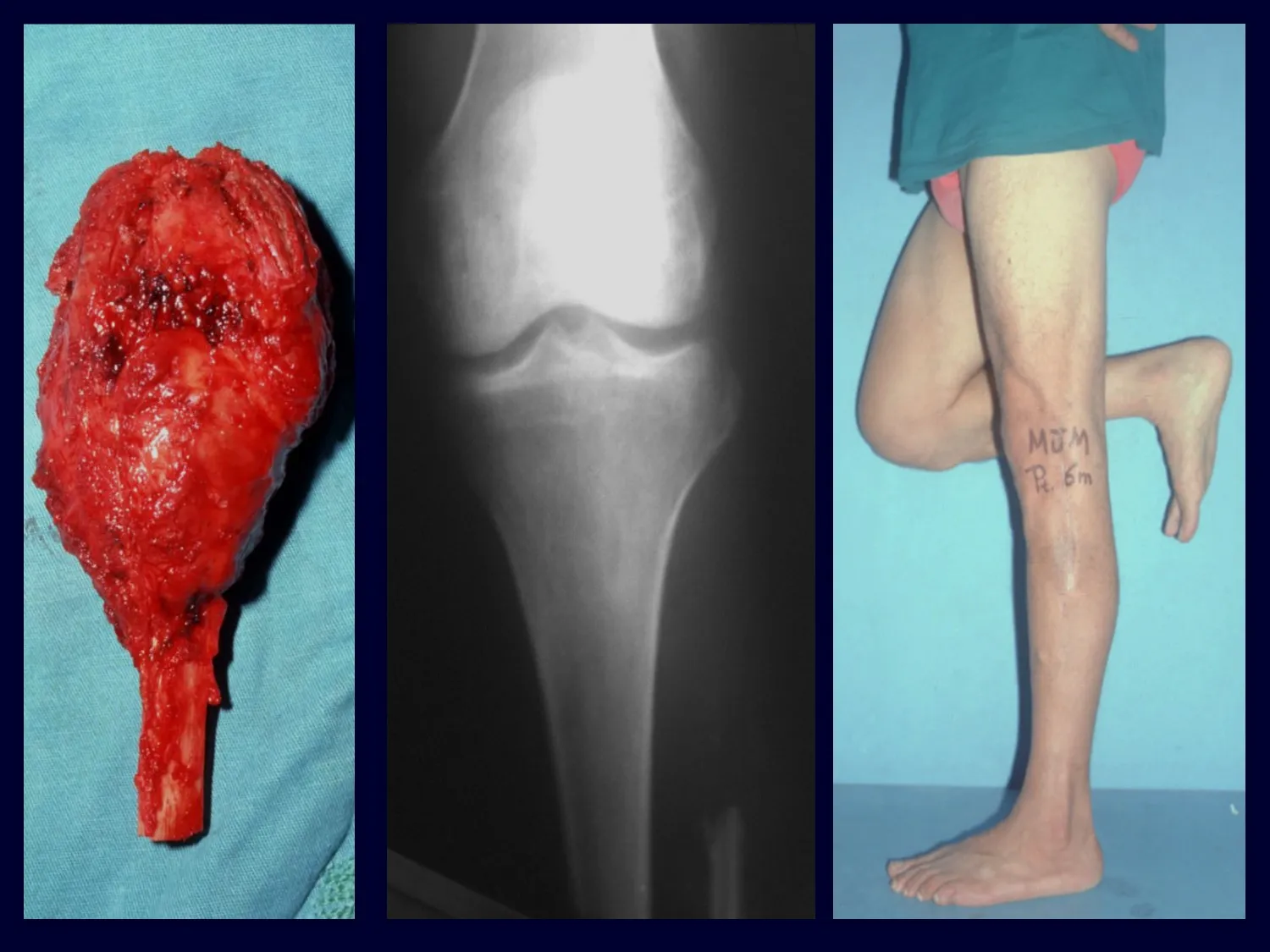
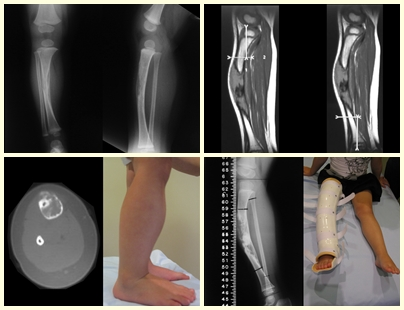
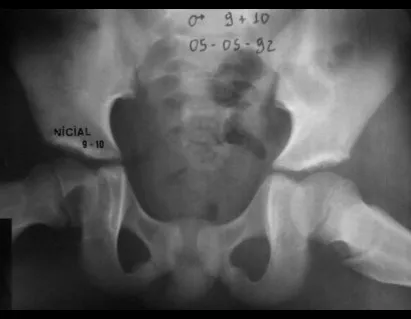
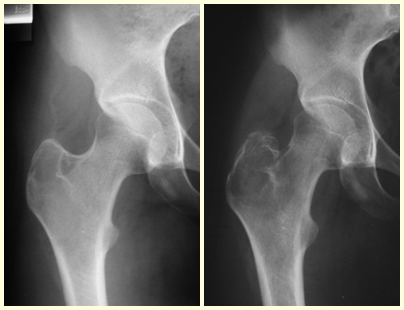
Radiológicamente se describe como una lesión epifisaria caracterizada por rarefacción ósea, inicialmente excéntrica, respetando inicialmente los límites corticales. A medida que avanza, puede producirse rotura cortical y afectación articular (fig. 27).
Histológicamente, las células gigantes y el estroma son los elementos más importantes de este tumor. Se caracteriza por tener numerosas células gigantes que se asemejan a los osteoclastos en un estroma de células fusiformes.
Los principales diagnósticos diferenciales clínicos, radiológicos y anatomopatológicos son el quiste óseo aneurismático, el osteosarcoma teleangectásico y el condroblastoma.
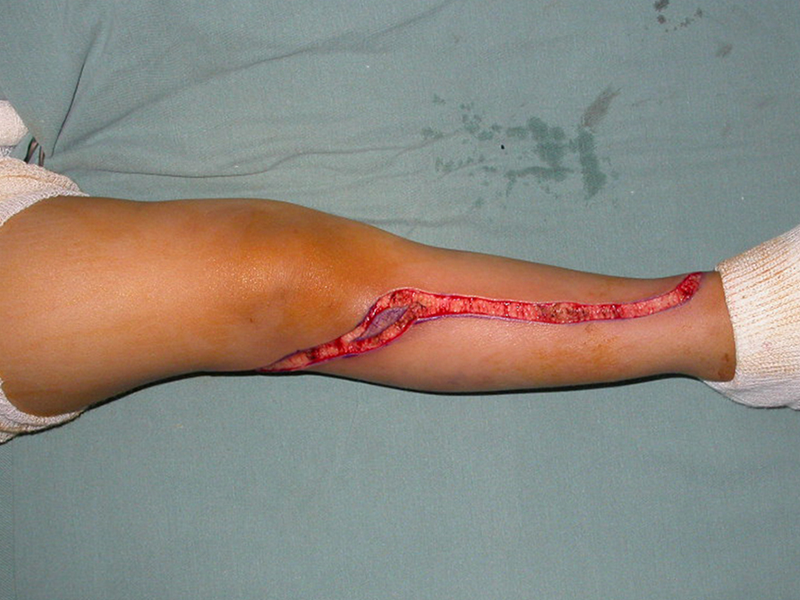
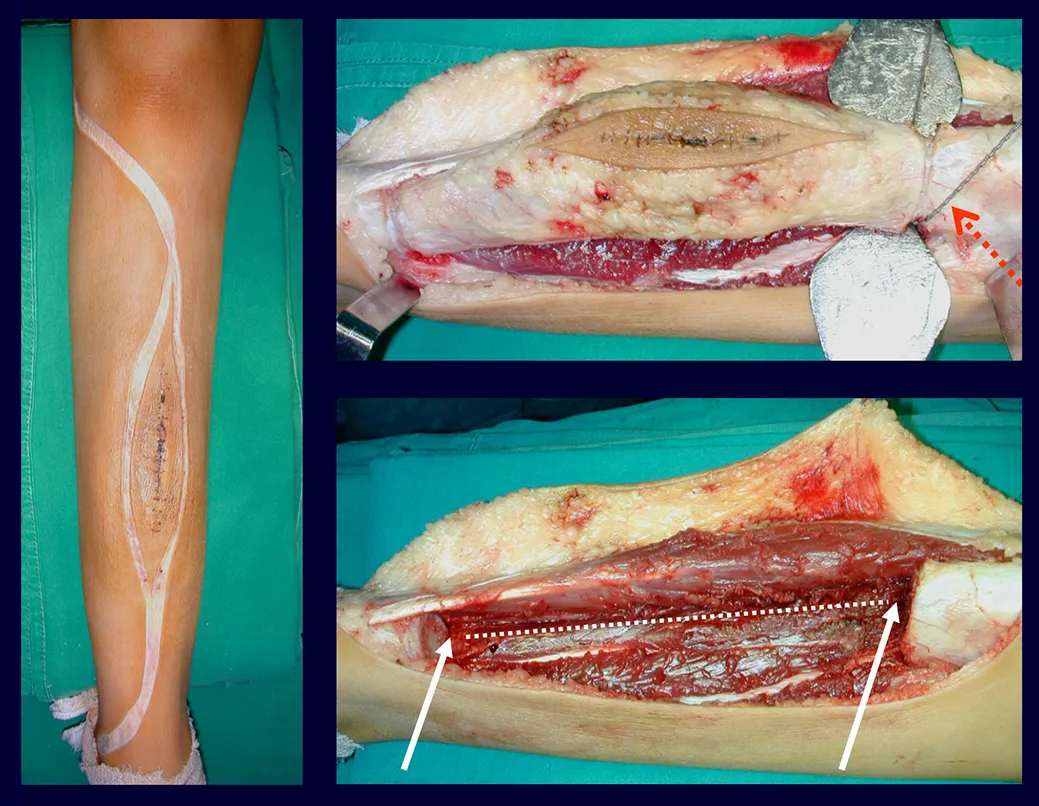
El tratamiento del tumor de células gigantes está actualmente bien establecido. Siempre que sea posible se debe optar por la resección segmentaria de la lesión, con margen de seguridad oncológica tanto en hueso como en tejidos blandos. De esta forma el cirujano brindará la mayor oportunidad de curación, sin riesgo de recurrencia.
Sin embargo, en regiones donde la resección segmentaria no es factible, como la columna cervical por ejemplo, se debe realizar el legrado endocavitario más sensato posible y complementarlo con terapia adyuvante como el láser de CO 2 , el fenol diluido en alcohol al 4%, el nitrógeno líquido y la electrotermia. El metilmetacrilato tiene un efecto adyuvante bajo y, cuando se utiliza para rellenar la cavidad, debe ir precedido de una de las terapias mencionadas.
En el pasado, el curetaje tenía altas tasas de recurrencia debido a la falta de grandes aberturas para una limpieza eficaz y la falta de uso de adyuvantes locales. Hoy en día, cuando está indicado el legrado endocavitario, se recomienda crear una ventana ósea grande para permitir al cirujano una visión amplia de la cavidad. Complementamos también este curetaje con el uso de un bisturí eléctrico (electrotermia).
Esta técnica electrotérmica es muy efectiva porque con la punta curva del bisturí podemos llegar a zonas de más difícil acceso. Este bisturí, además de cauterizar, también realiza un complemento al legrado, ya que aquellas células tumorales, que quedan en las pequeñas “cavidades” de la pared ósea, se destruyen y se desprenden, permitiendo su eliminación más fácil.
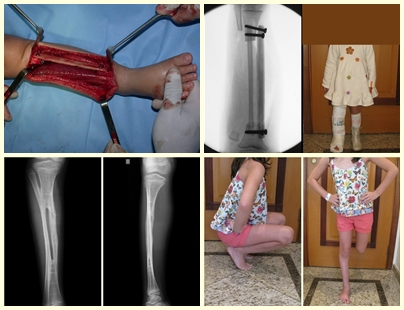
Complementamos nuestro curetaje, tras utilizar electrotermia, con el fresado de la cavidad. Para ello utilizamos el Lentodril , con una fresa dental esférica.
En la región de la rodilla (extremo distal del fémur y extremo proximal de la tibia), el sitio de mayor incidencia de CGT, recomendamos frecuentemente legrado endocavitario, seguido de electrotermia y fresado con Lentodril . Esto se debe a que la resección segmentaria de esta región implicaría artrodesis o sustitución por endoprótesis o injertos homólogos. La artrodesis de la articulación de la rodilla supone la mayor limitación para el paciente y debe evitarse. Los recambios en pacientes jóvenes pueden acarrear problemas en un futuro próximo y su indicación debe ser prudente.
Por ello, recomendamos en primer lugar la terapia más conservadora: – legrado seguido de terapia adyuvante en esta región, excepto en casos avanzados, con destrucción importante de la estructura ósea, en los que puede verse comprometido tanto la función como el control local de la enfermedad.
Queda un breve comentario sobre el llenado de la cavidad tratada. Esto se puede realizar con injerto óseo autólogo o heterólogo o con metacrilato de metilo, cada uno de ellos tiene sus ventajas y desventajas.
El metacrilato de metilo permite visualizar fácilmente posibles recurrencias, es fácil de usar y permite una carga más temprana, sin embargo no es una solución biológica y pueden ocurrir fracturas en la región.
El injerto óseo es una solución biológica y definitiva, sin embargo dificulta visualizar una posible recidiva temprana, que puede confundirse con reabsorción/integración del injerto y aún requiere seis meses en promedio para su carga completa. El injerto homólogo tiene un período de integración más largo, no siempre está disponible, pero por otro lado acorta el tiempo quirúrgico. El injerto autólogo tiene la ventaja de una biocompatibilidad y una integración más rápida, pero prolonga el tiempo quirúrgico.