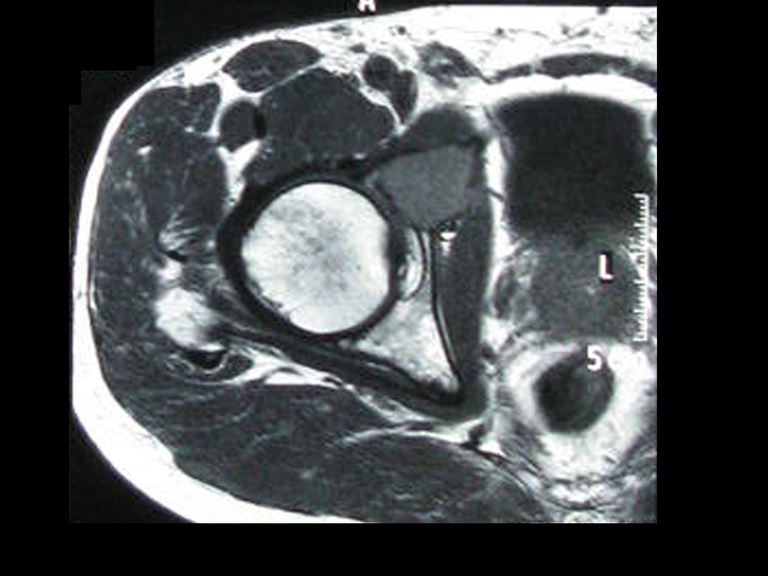
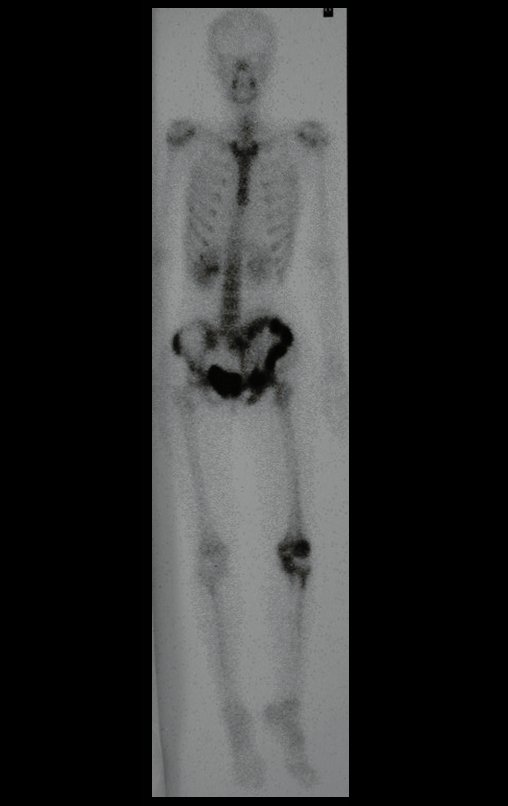
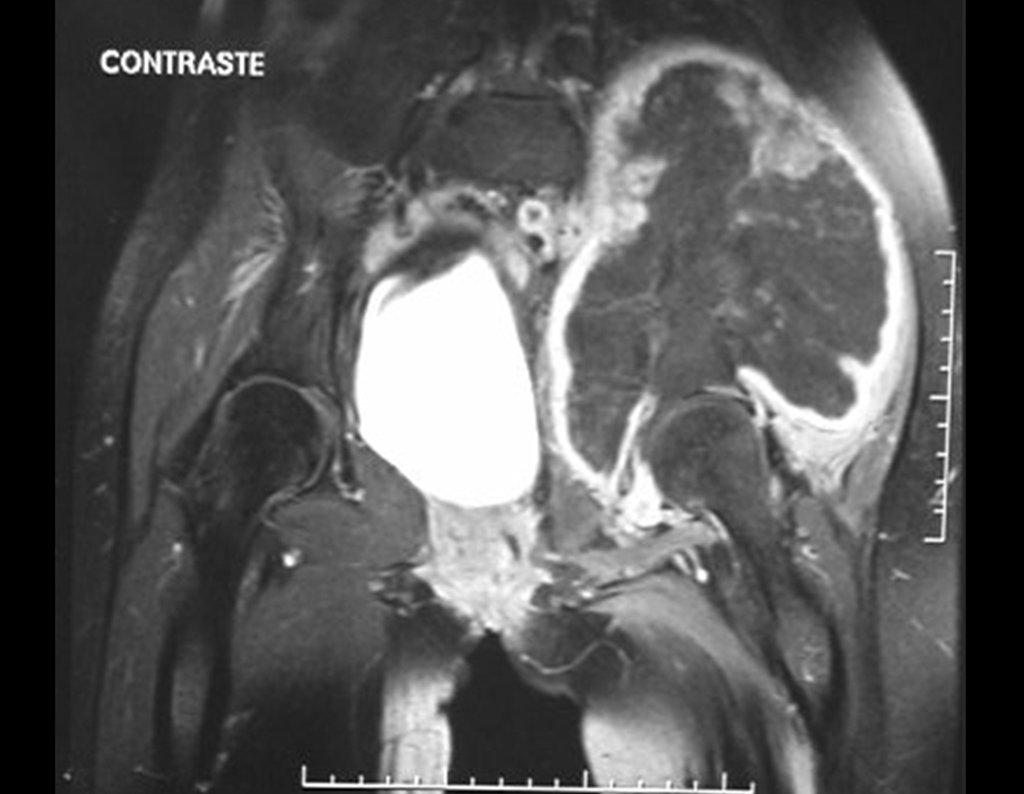
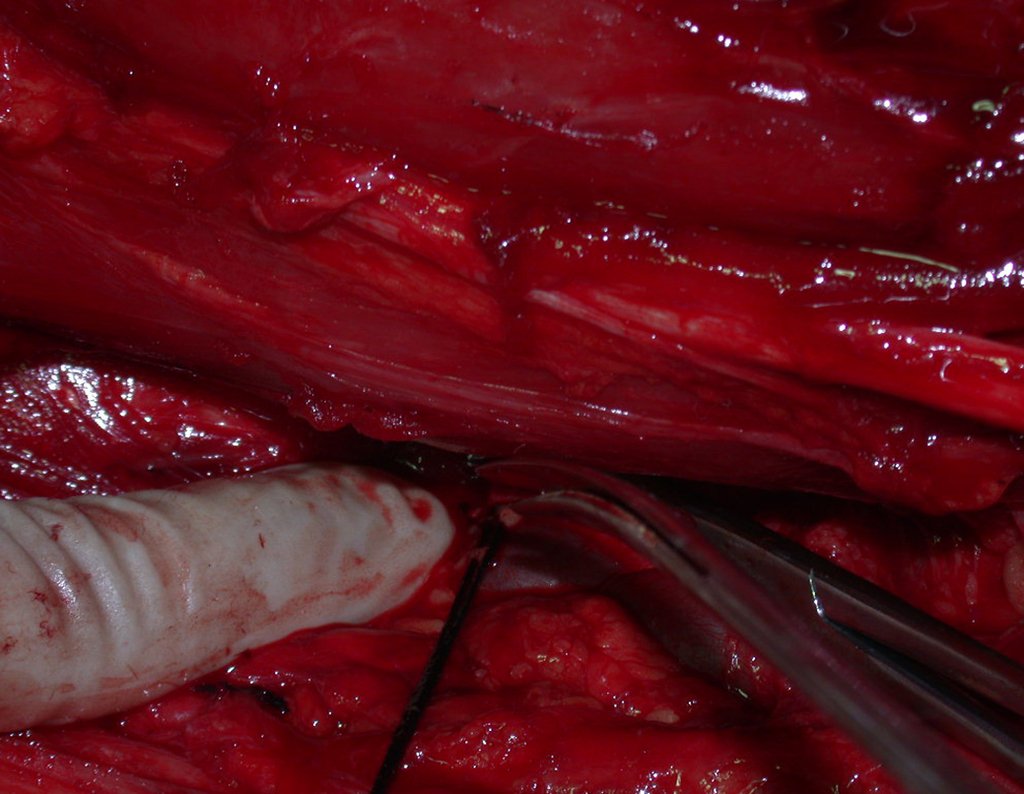
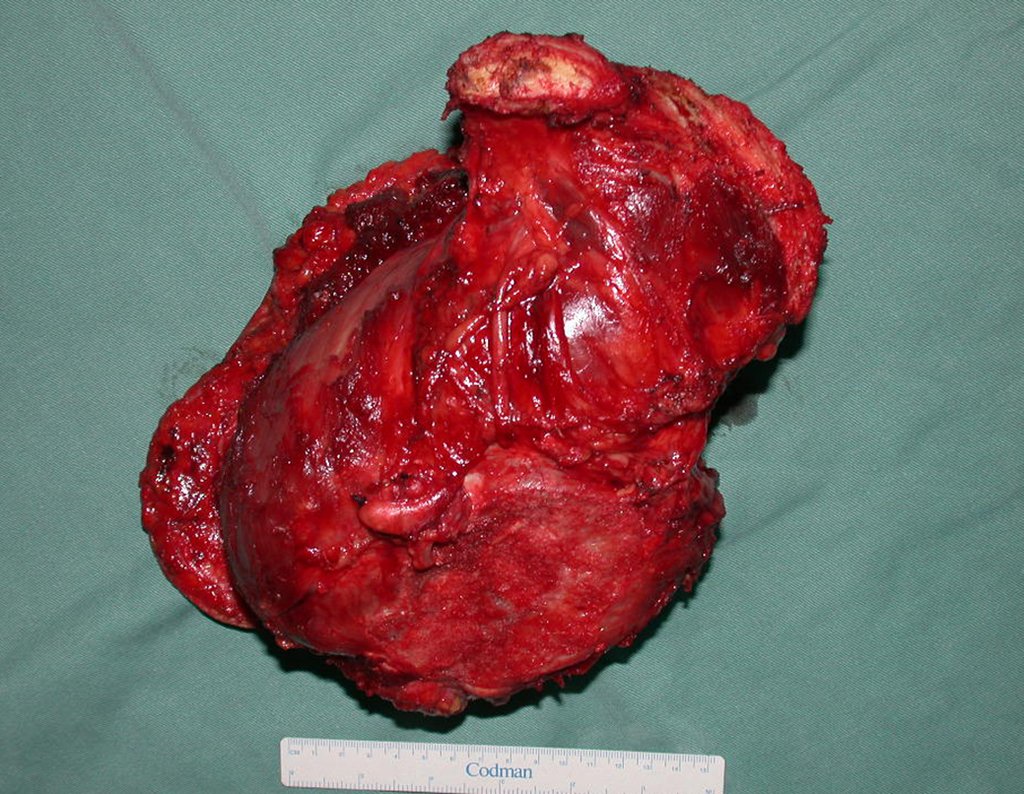
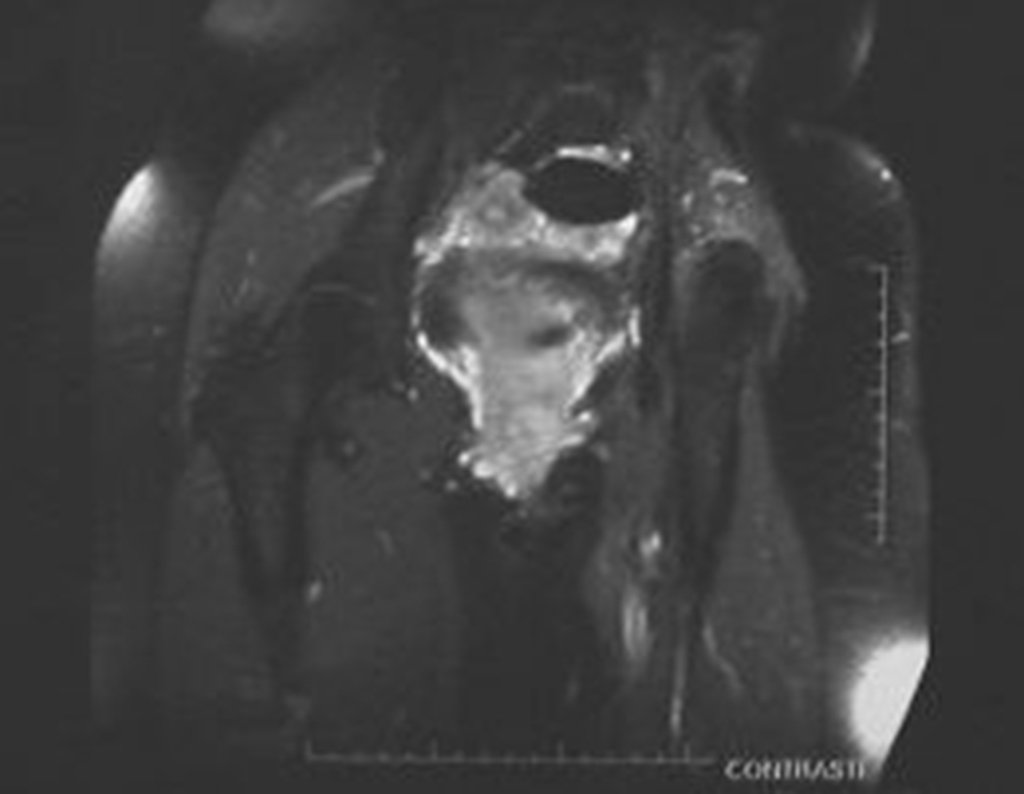
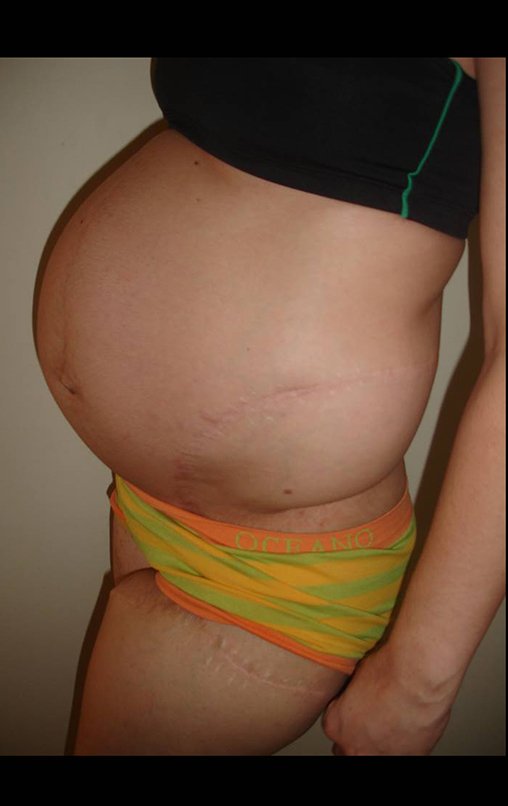
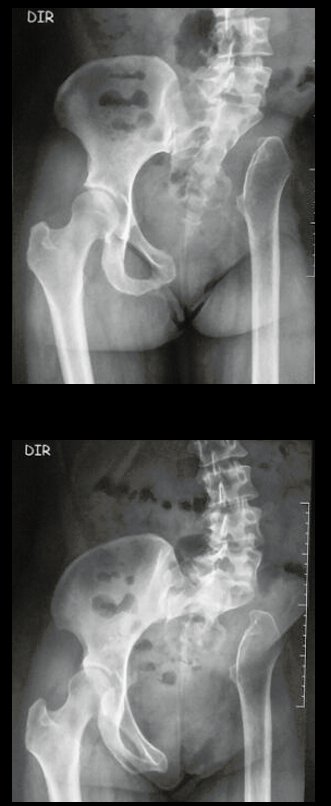
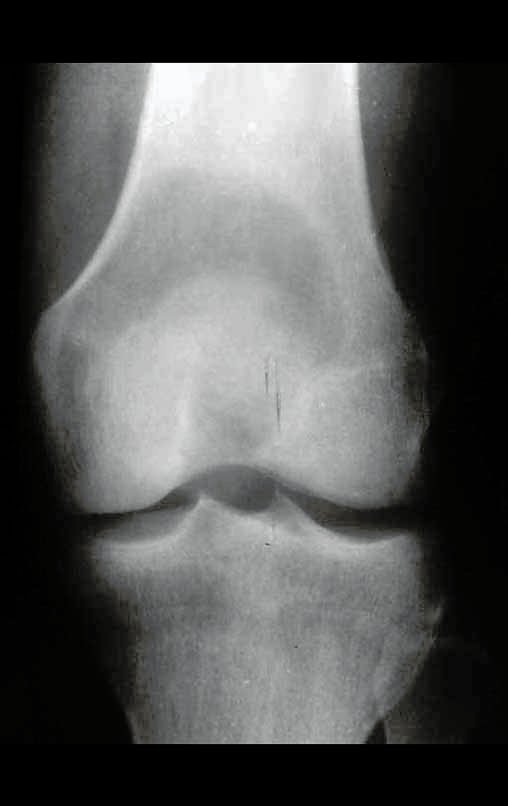
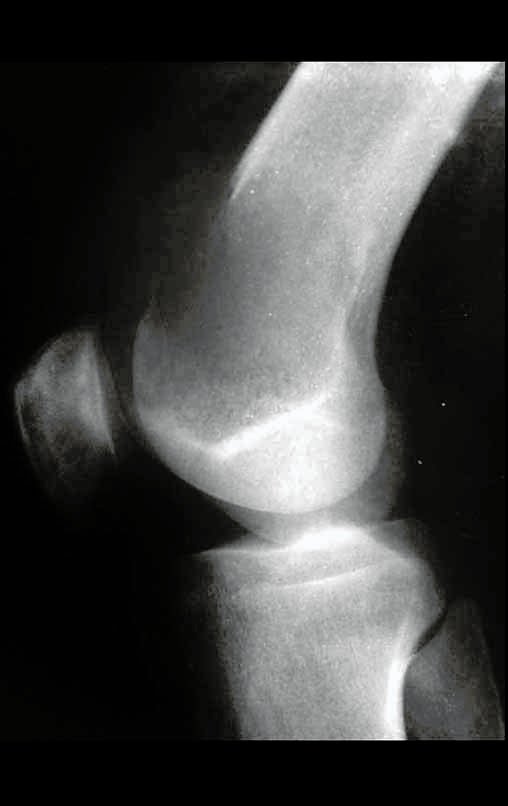
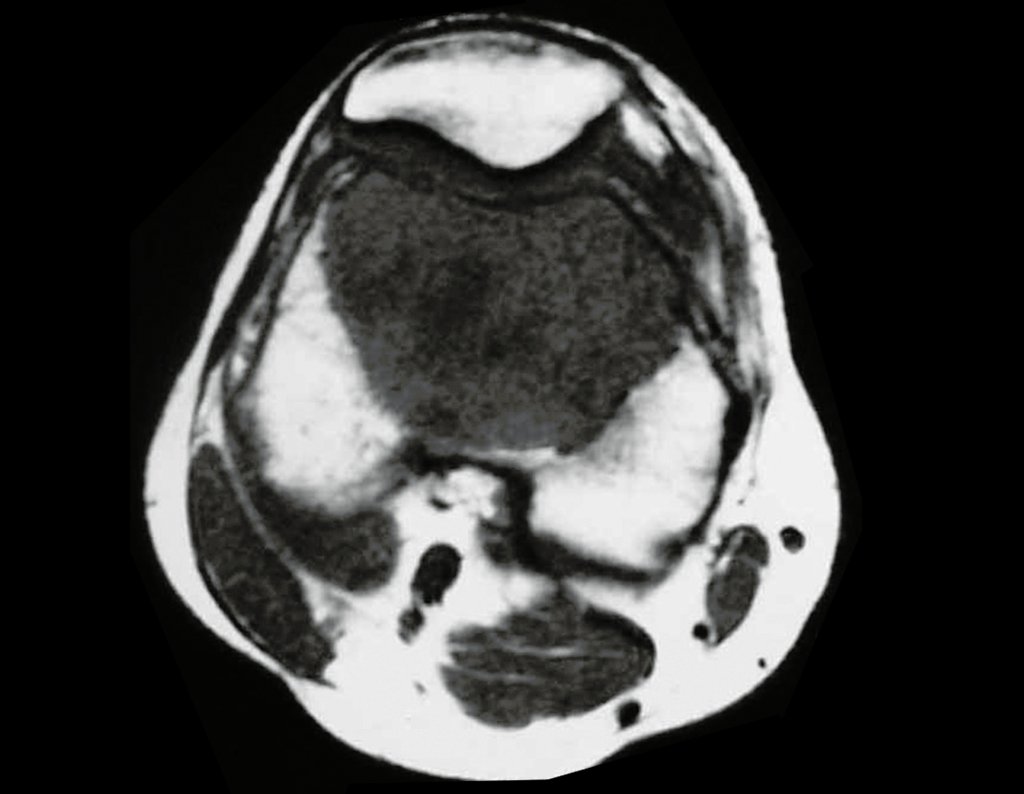
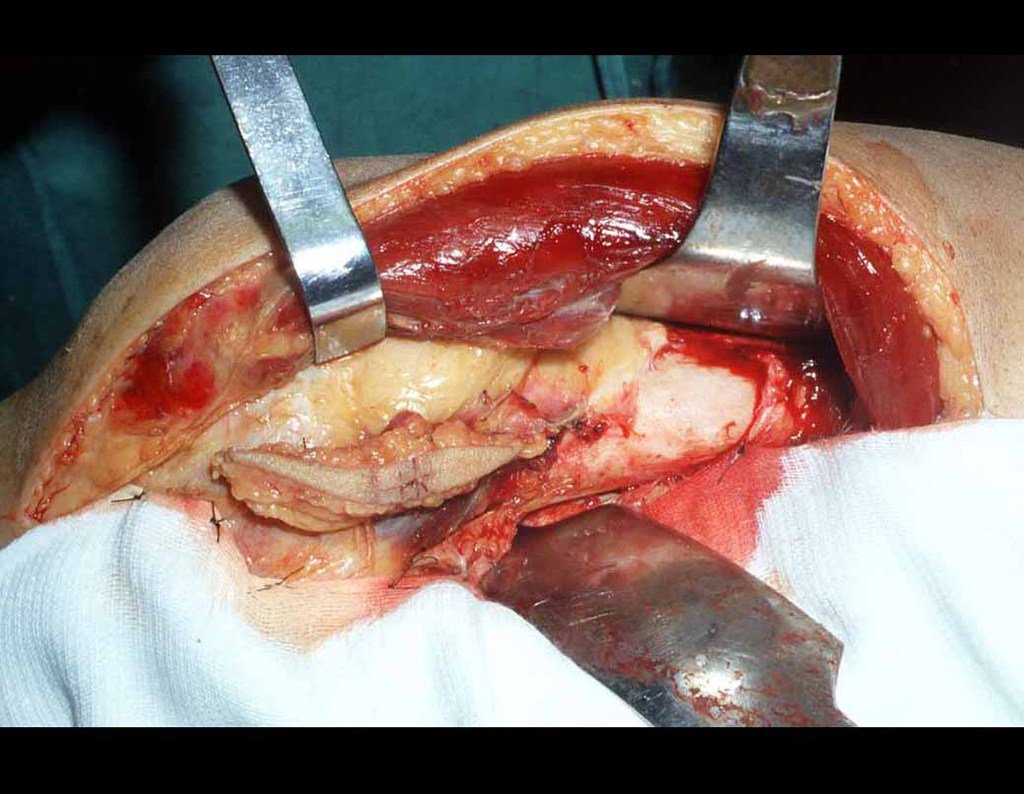
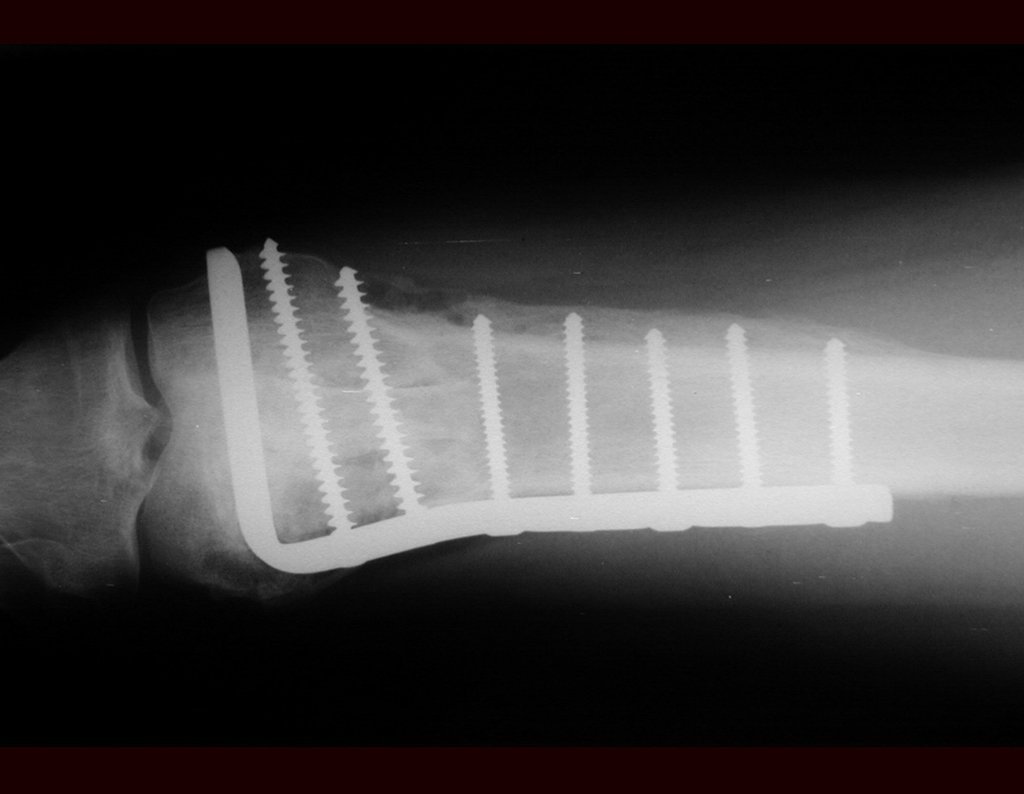
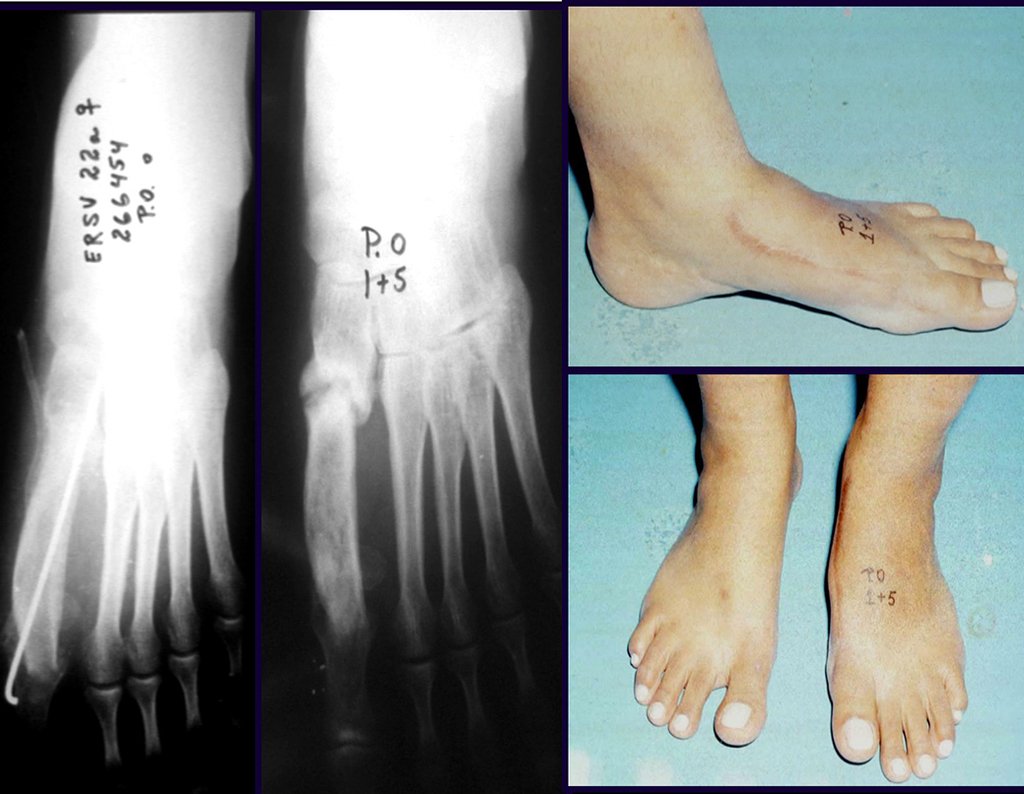
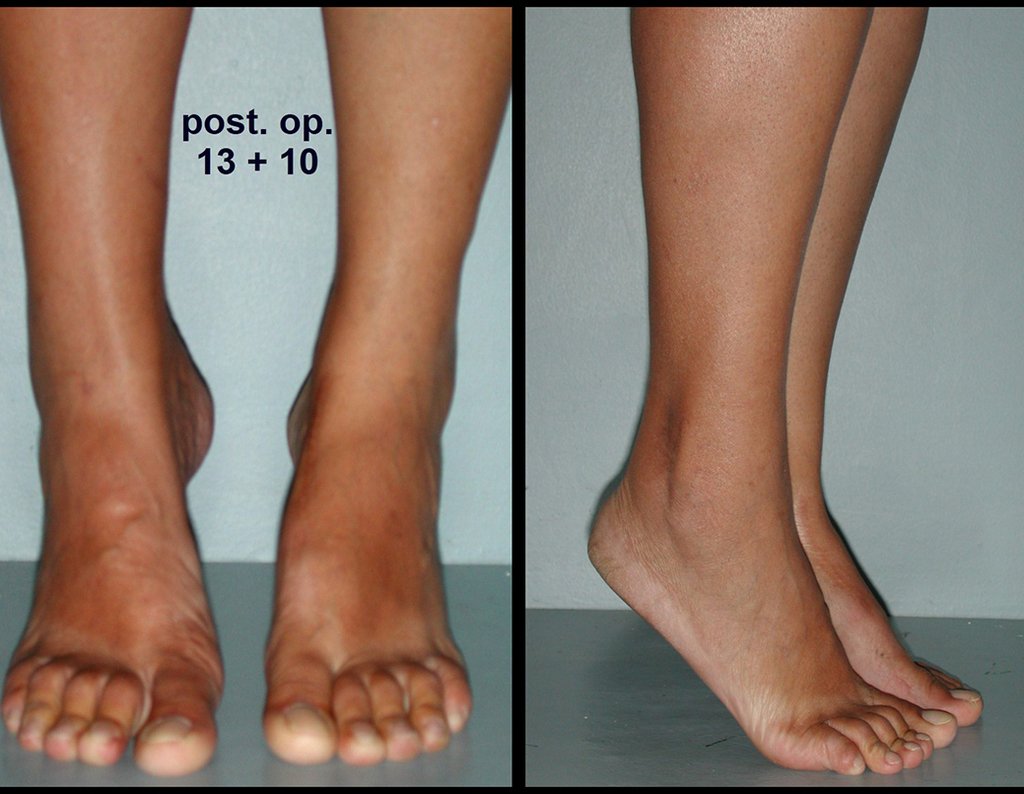
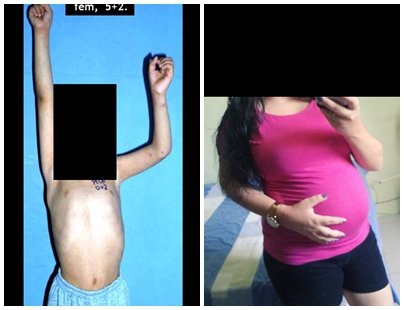
Diagnóstico Diferencial:
Presenta diagnóstico diferencial con la miositisosificante, o fibroma condromixóide, o T.C.G., o linfoma no Hodgkin6,23,29 y con un quiste óseo aneurismatoco, por ser de carácter multiloculado. Histológicamente, el subtipo cortical asemeja al condroma, al osteocondroma, al condroblastoma y al osteosarcoma de superficie16.
El condrosarcoma de células claras tiene condrocitos malignos con citoplasma claro, células gigantes tipo osteoclastos y formación de hueso reactivo intralesional causando confusión con osteosarcoma.
El osteosarcomamesenquimal es formado por islas de cartílago hialino bien diferenciado circundado por laminas de células pequeñas y redondas, que asemejan al hemangiopericitoma y sarcoma de Ewing14.
El condroma central de los huesos largos, o condrosarcoma y el infarto óseo son muchas veces de difícil diferenciación, necesitando acompañamiento clínico y radiológico para evaluar la progresión de una lesión, antes de definir la conducta. La biopsia muchas veces no es definitiva para el diagnóstico12,23,29.
Tratamiento:
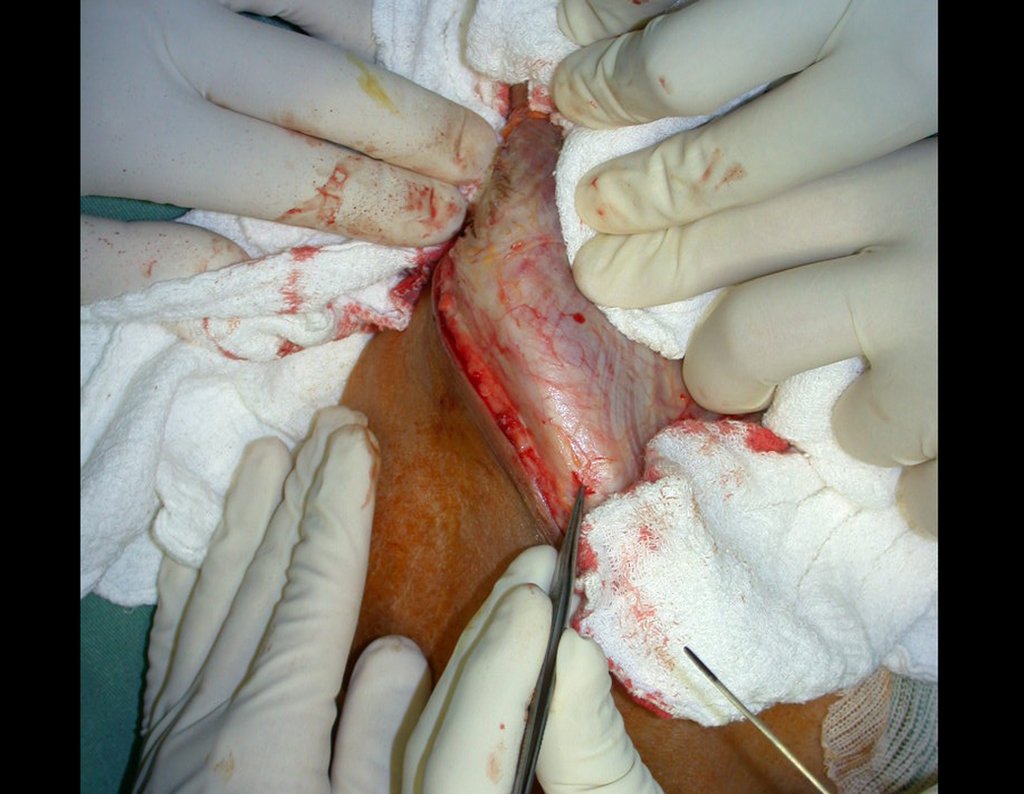
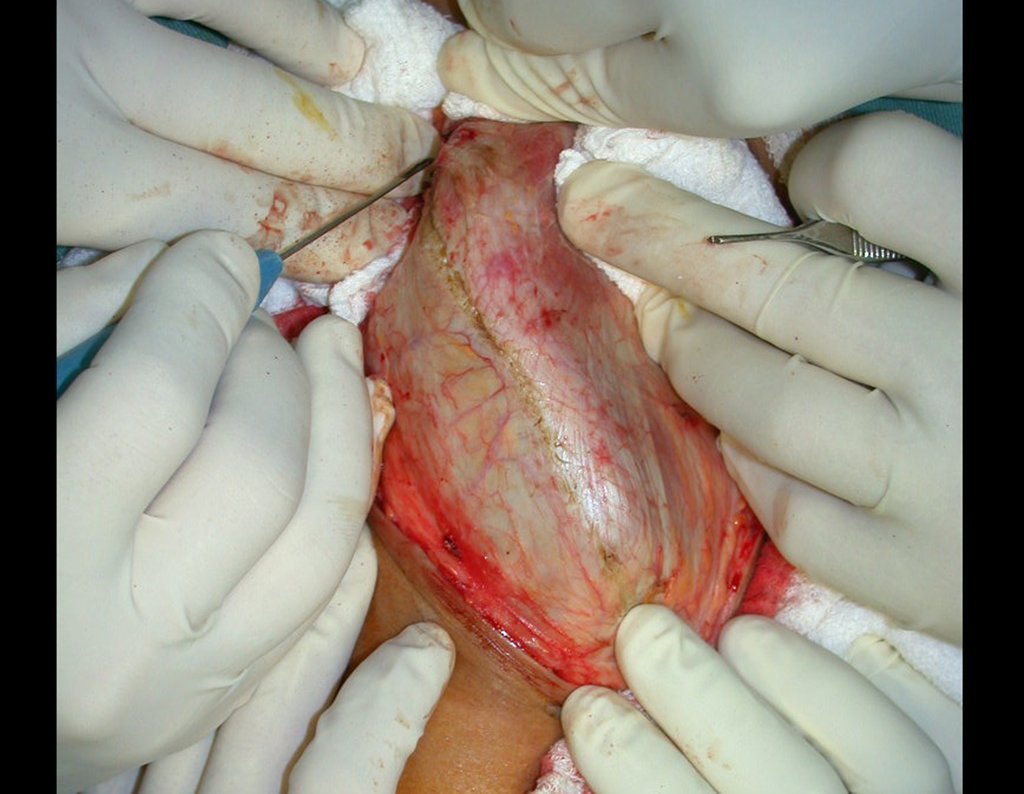
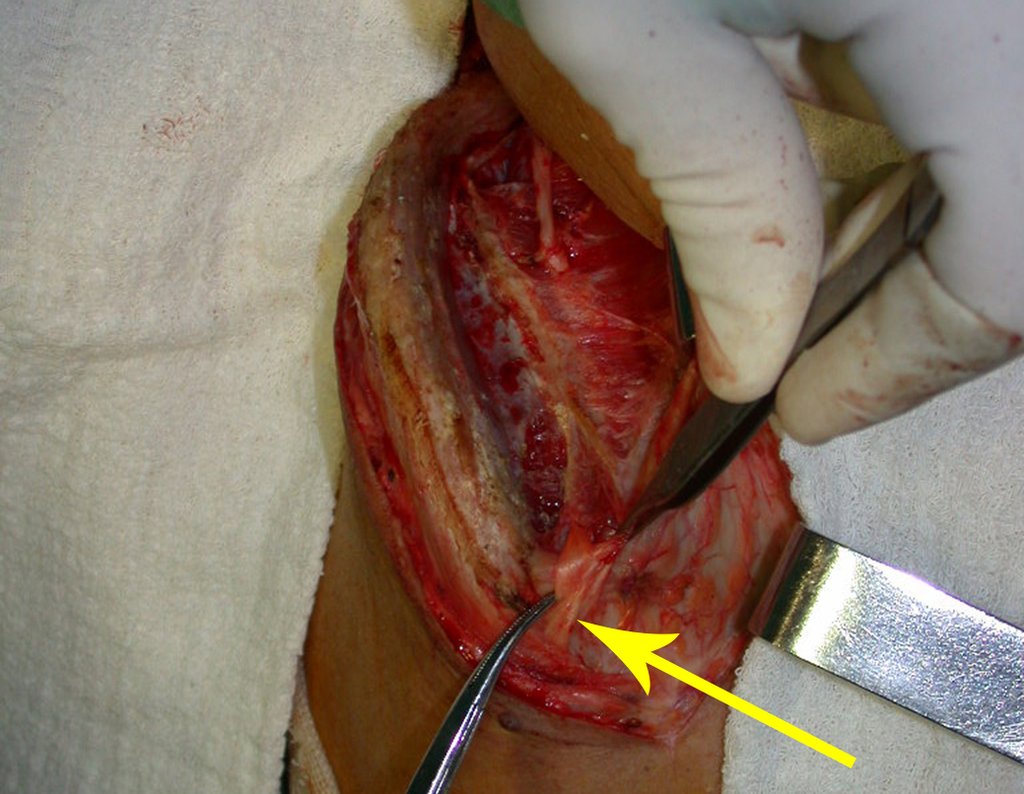
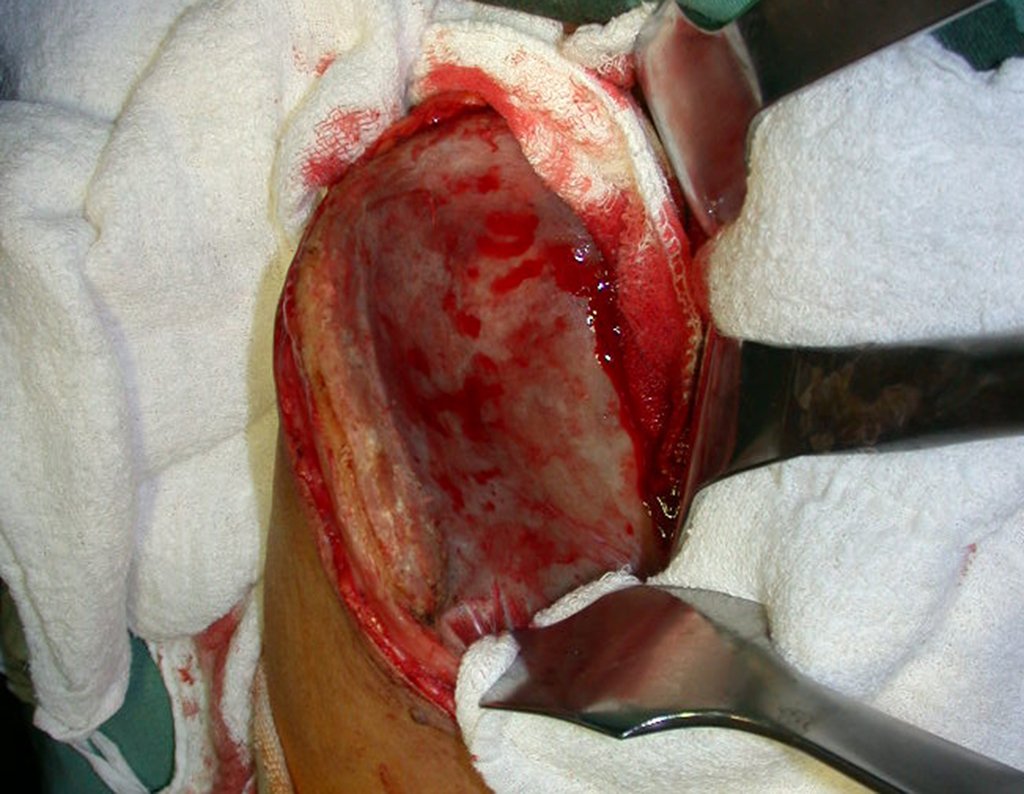
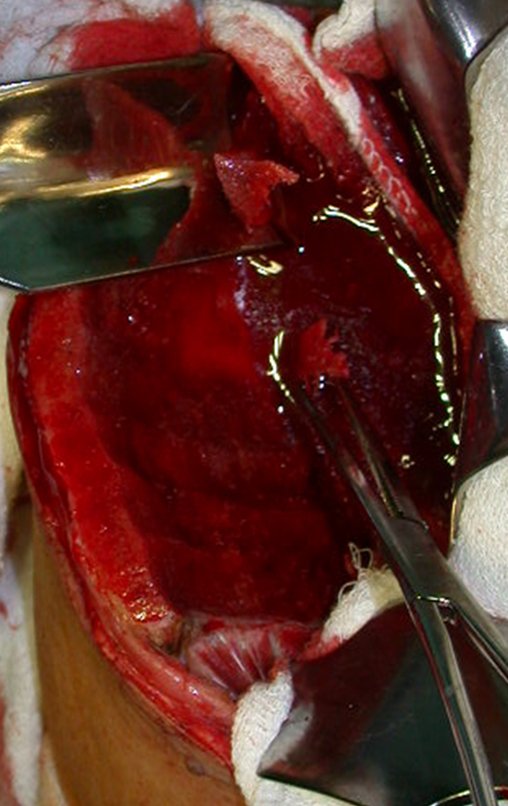
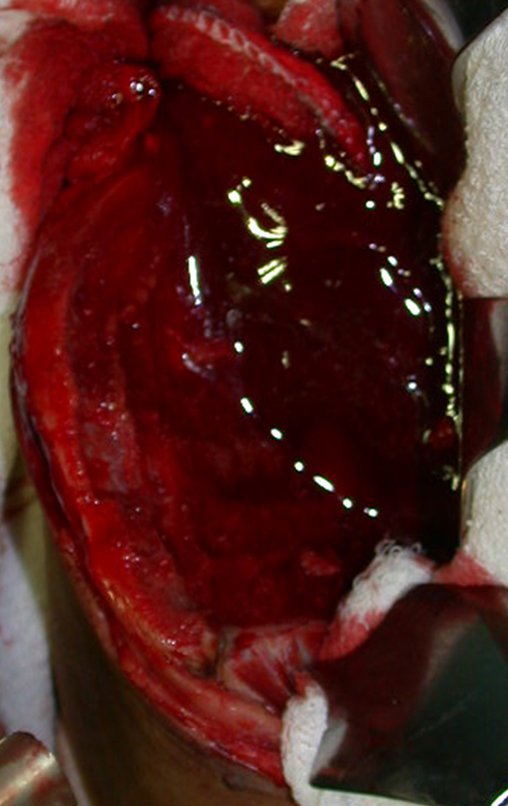
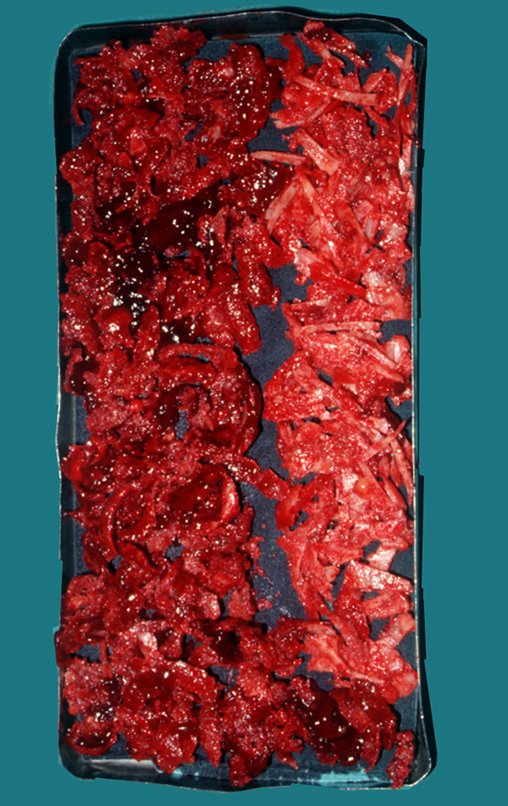
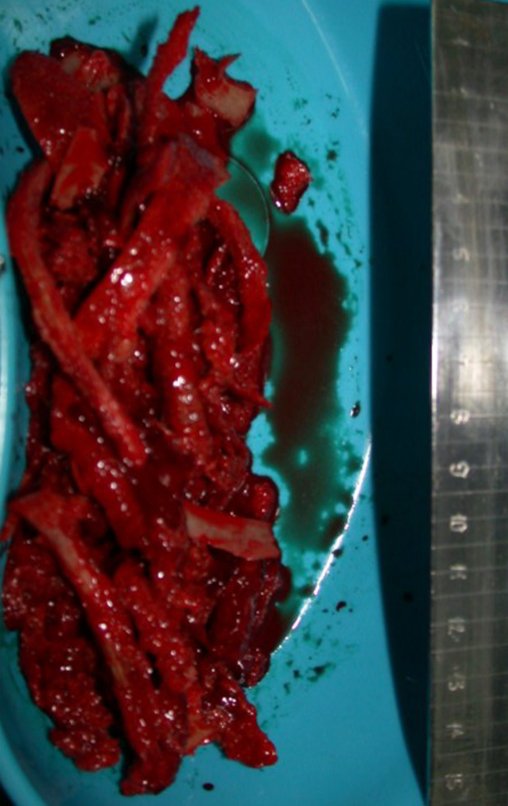
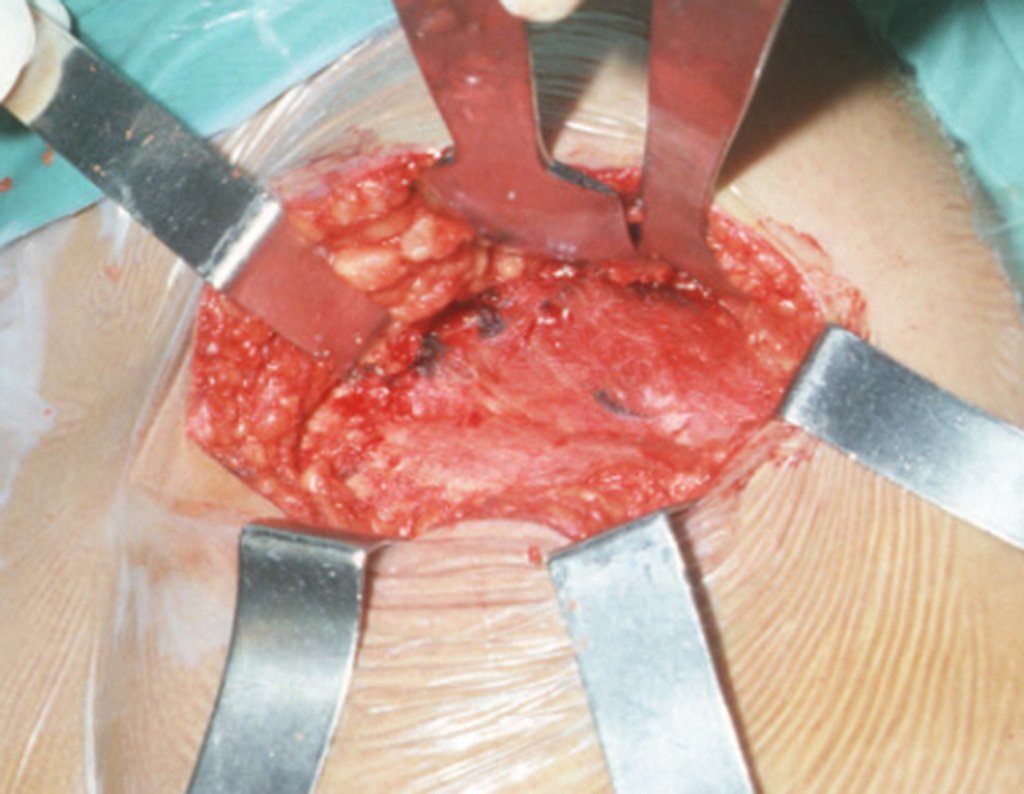
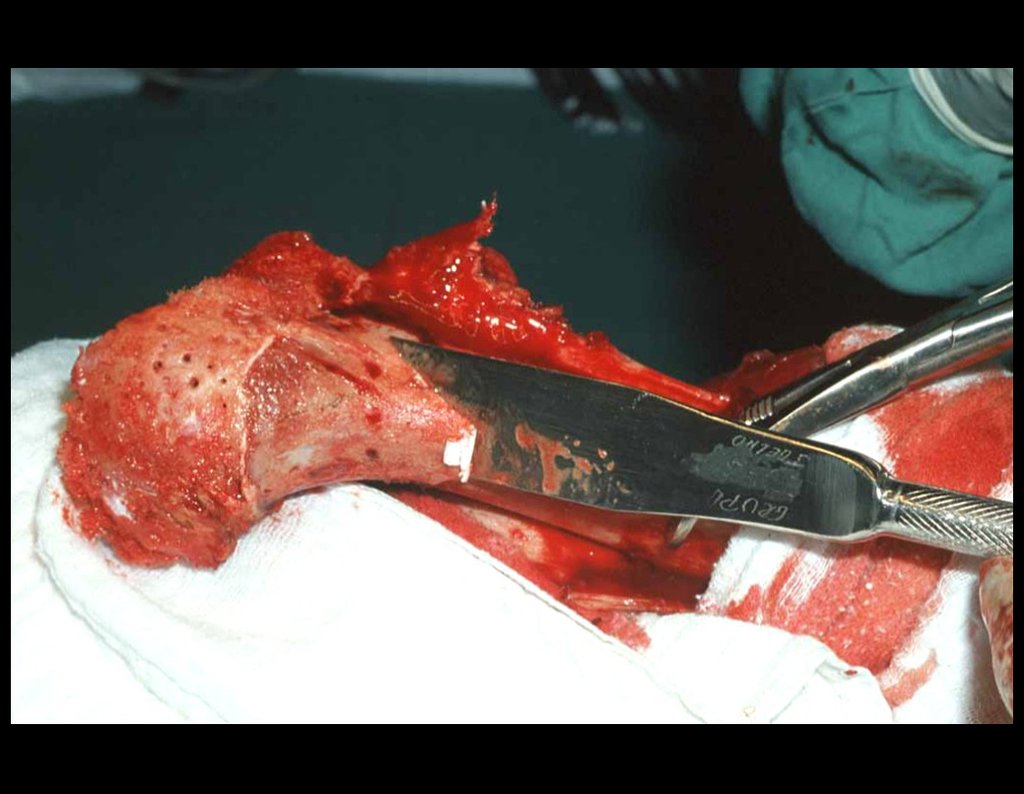
El tratamiento del condrosarcoma es quirúrgico25, dibiendo elegir una resección amplia, incluyendo el trayecto de la biopsia13,21.
La radioterapia es ineficaz6, no controla esta neoplasia. Para las lesiones de alto grado se puede discutir la indicación de quimioterapia utilizando el protocolo para sarcomas de grandes células, basado en antraciclicos9999. Para el condrosarcomamesenquimal, que presenta predominio de células pequeñas e indiferenciadas, cuando esta indicada la quimioterapia recae sobre el protocolo de tratamiento del tumor de Ewing.888
En ambos casos la respuesta a la quimioterapia suele ser mala6. El tratamiento de esta neoplasia debe ser particularizado para cada subtipo clínico:
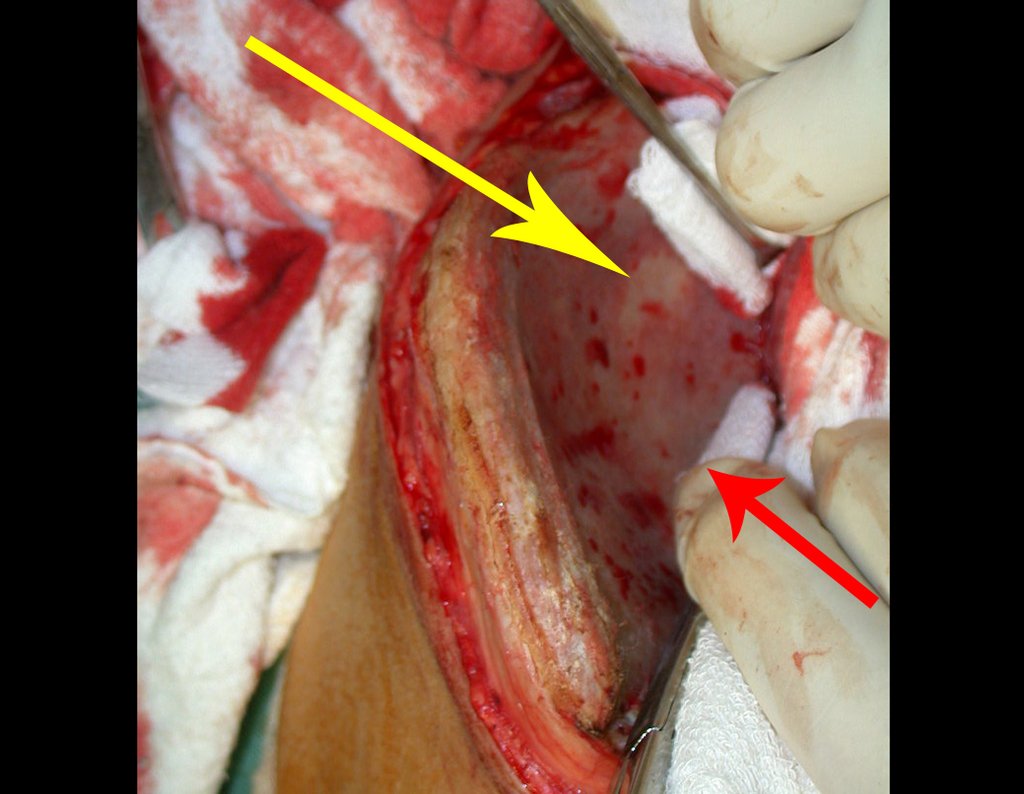
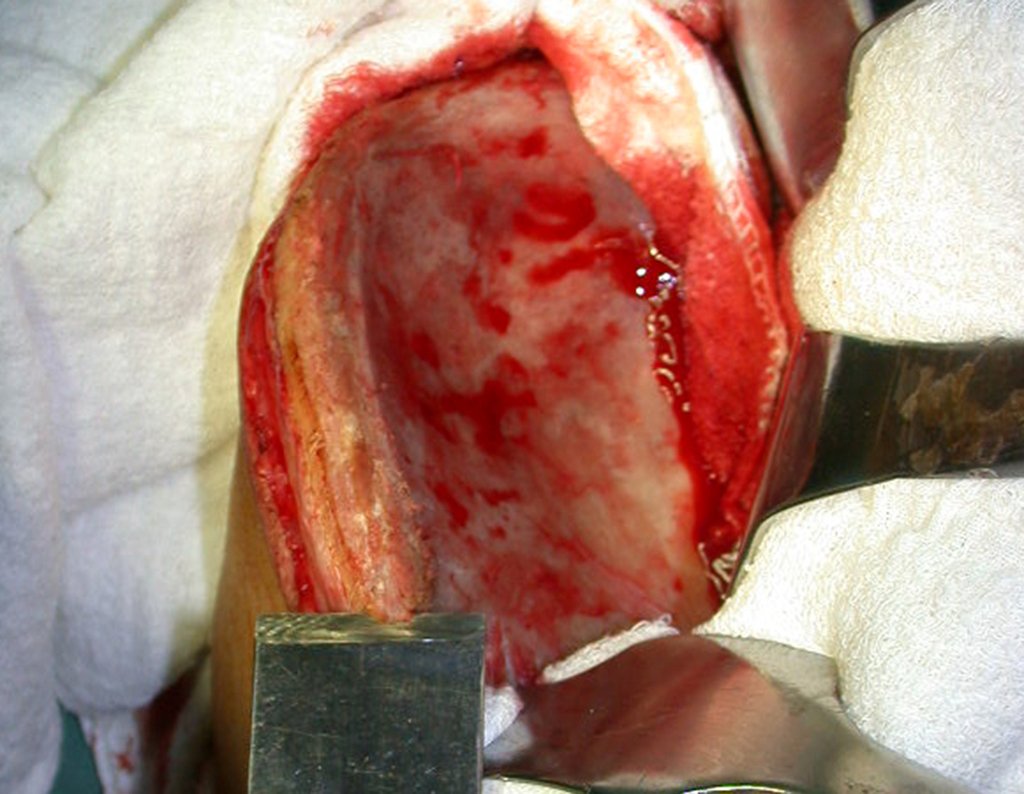
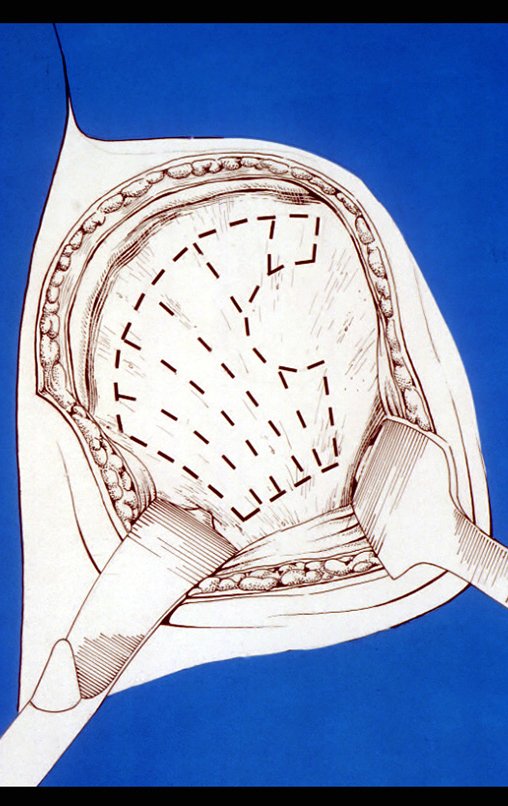
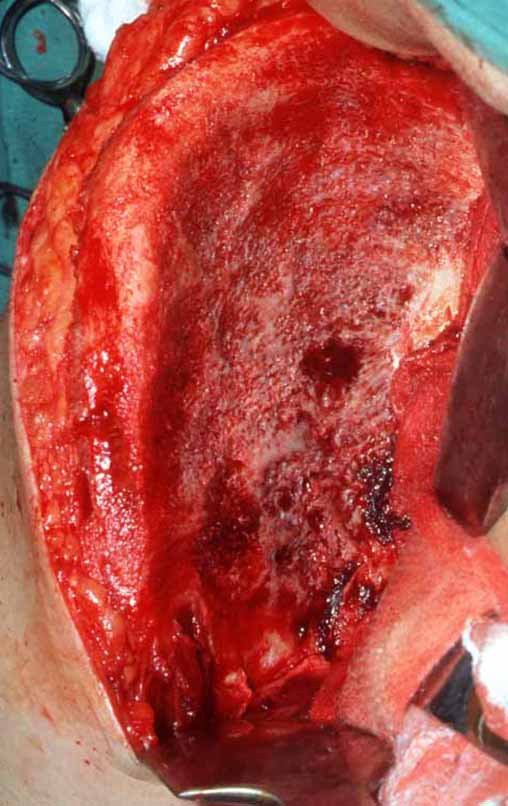
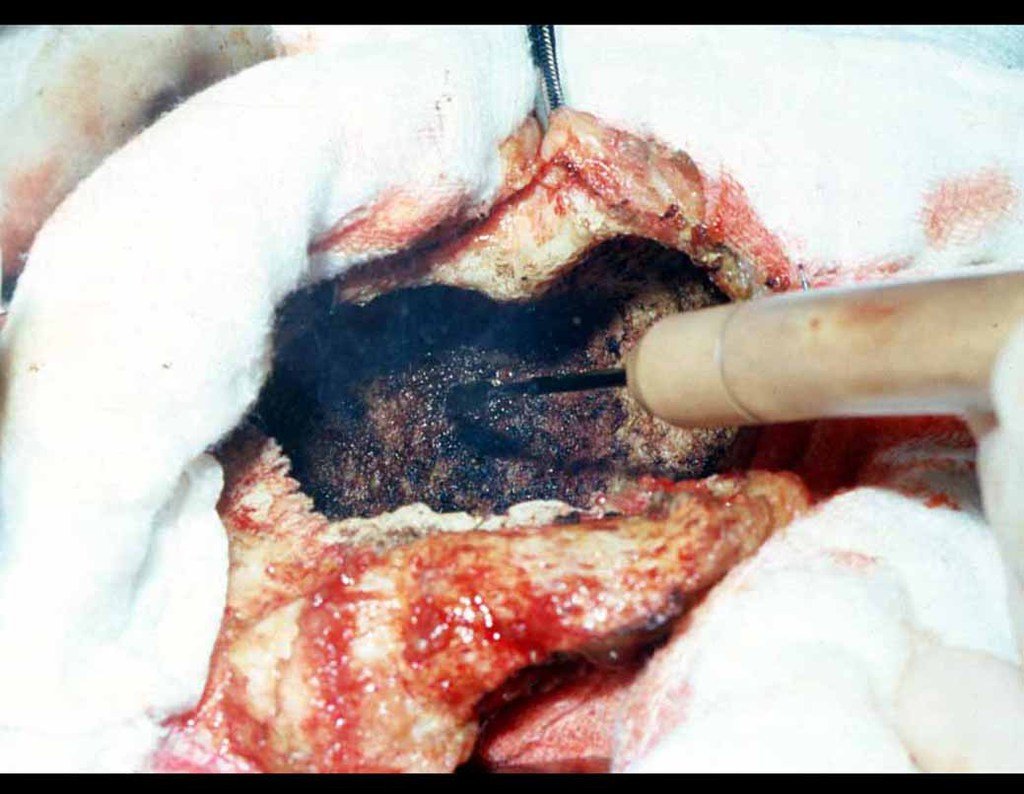
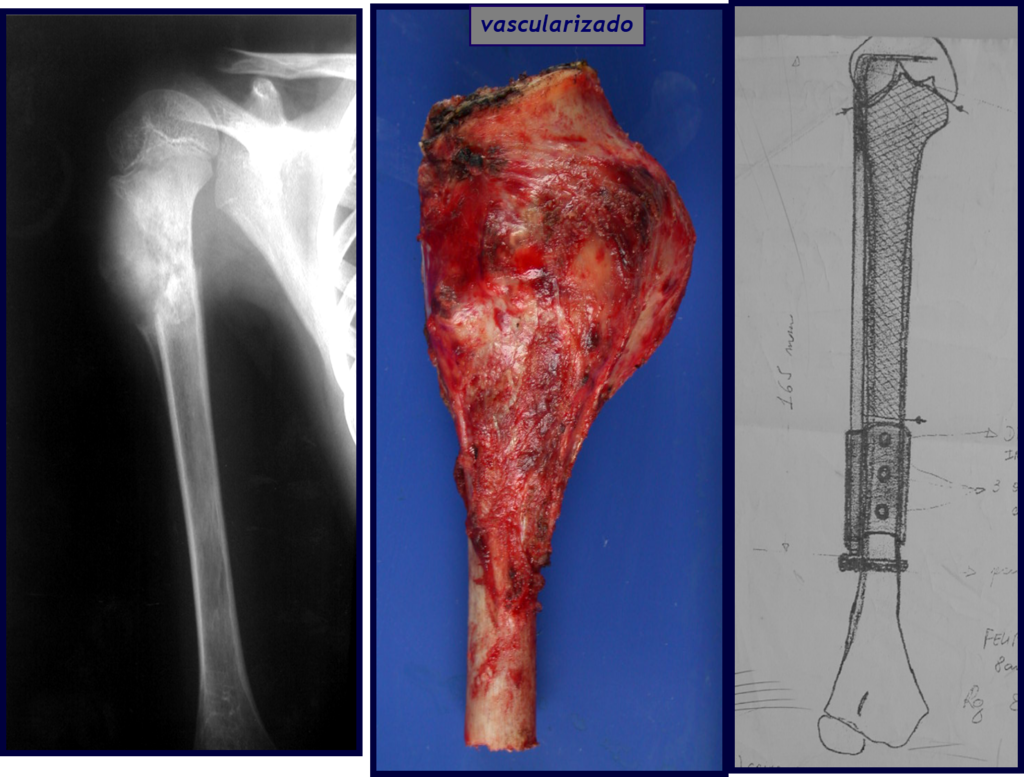
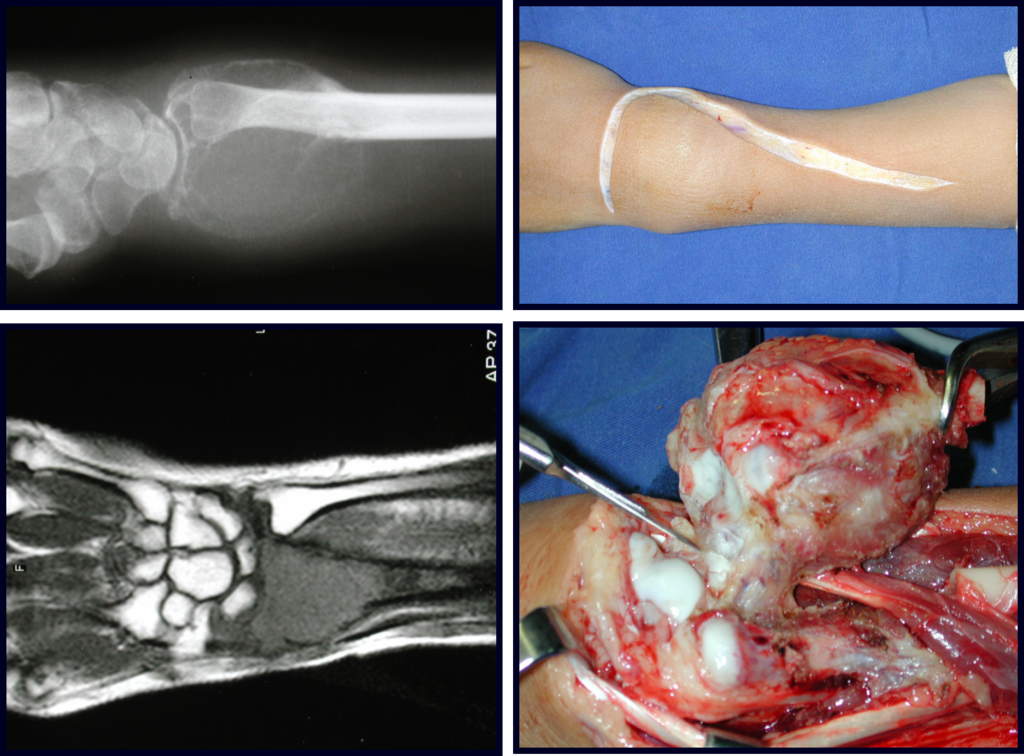
– Condrosarcoma central:presenta altos índices de cura con la cirugía adecuada, por lo tanto no se puede subestimar su tratamiento con curetajeintralesional seguido de métodos adyuvantes complementarios, sea con fenol, nitrógeno liquido, electrotermia o laser CO221.
De esta manera. En caso de dusa diagnóstica entre condroma y condrosarcoma de grado I es preferible observar la evolución de la lesión, pues es sabido que la biopsia no será concluyente, ya que el diagnóstico diferencial histológico entre condroma y condrosarcoma de grado I es difícil.
En algunos casos, estas lesiones pueden ser tratados con cirugía conservadoras sin la realización de biopsia previa21.
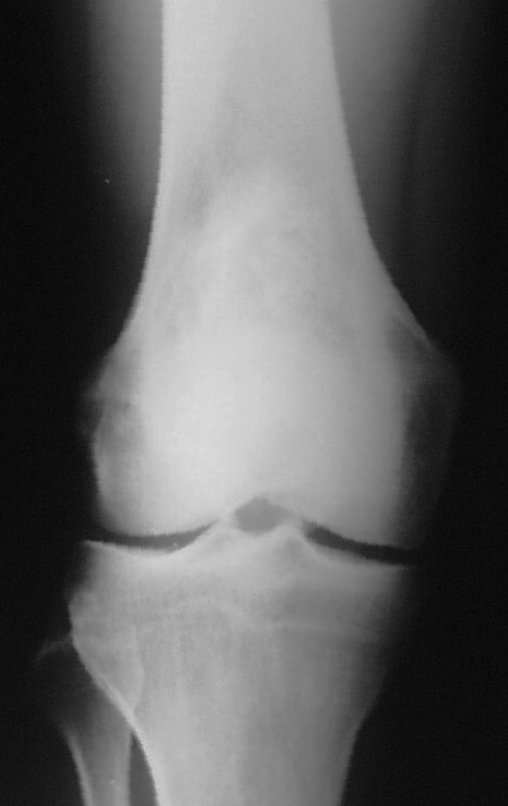
Cuando los exámenes de imagen: radiografía, TAC y RM, muestran una lesión central, sin erosión de la cortical interna, de grosor normal y sin dolor, se debe reevaluar dentro de tres meses, si no hay alteración se repite evaluación en seis meses y si permanece inalterado se programa reevaluaciones anuales.
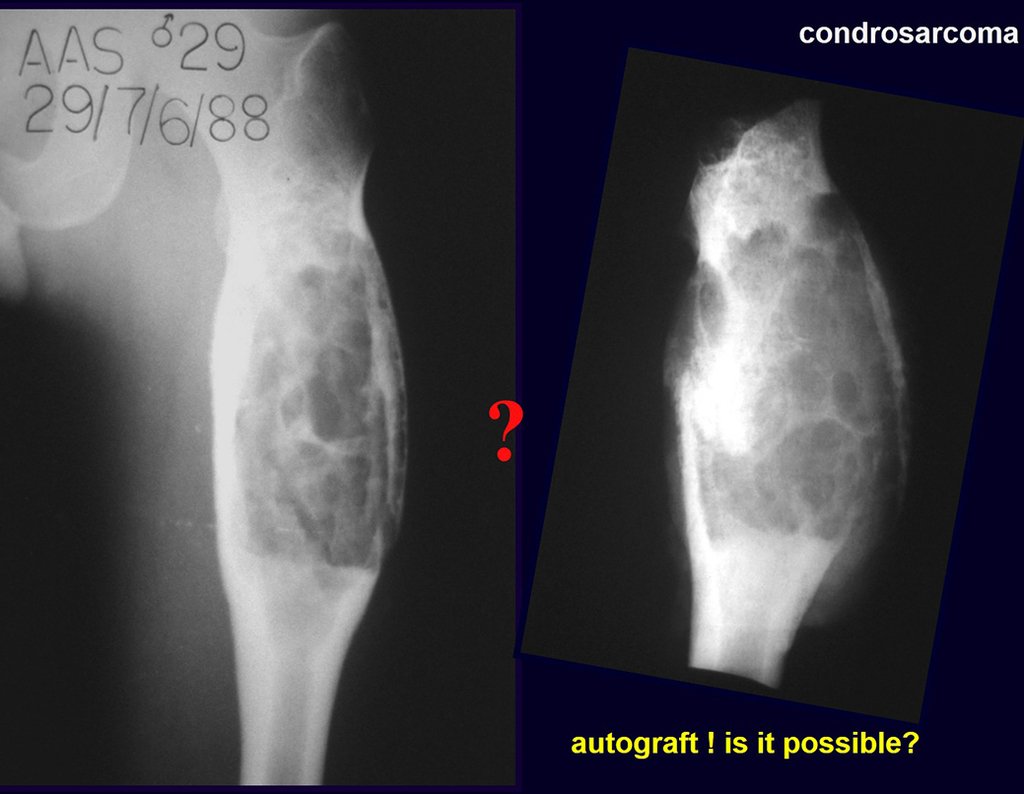
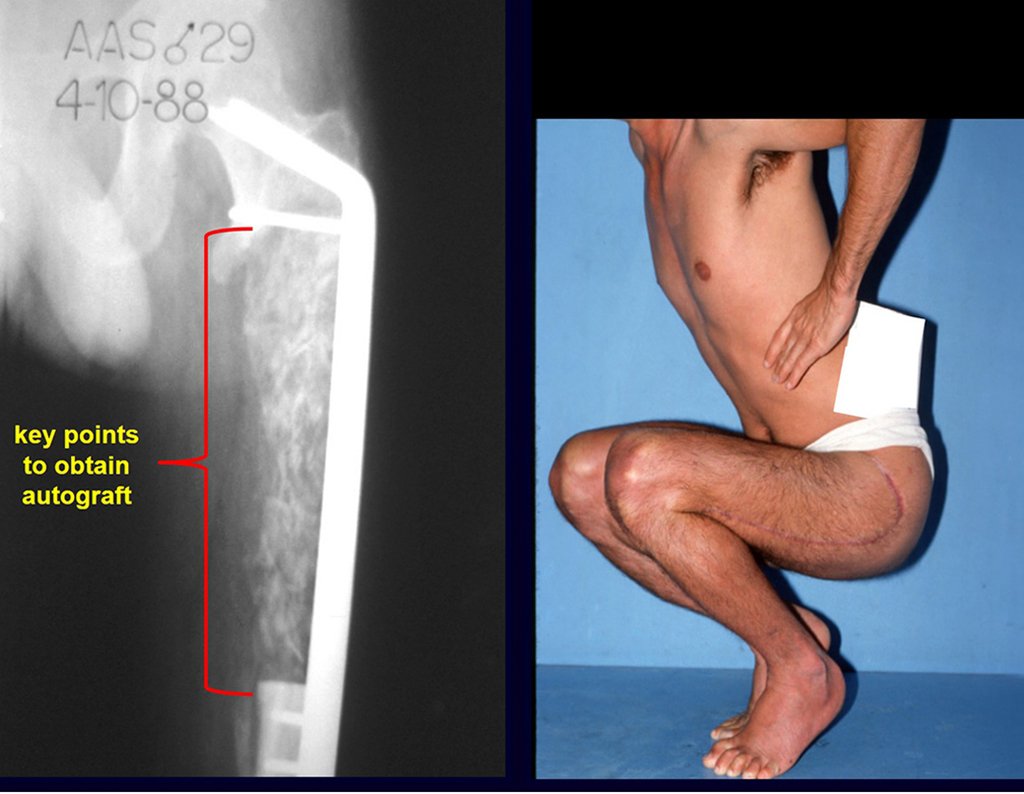
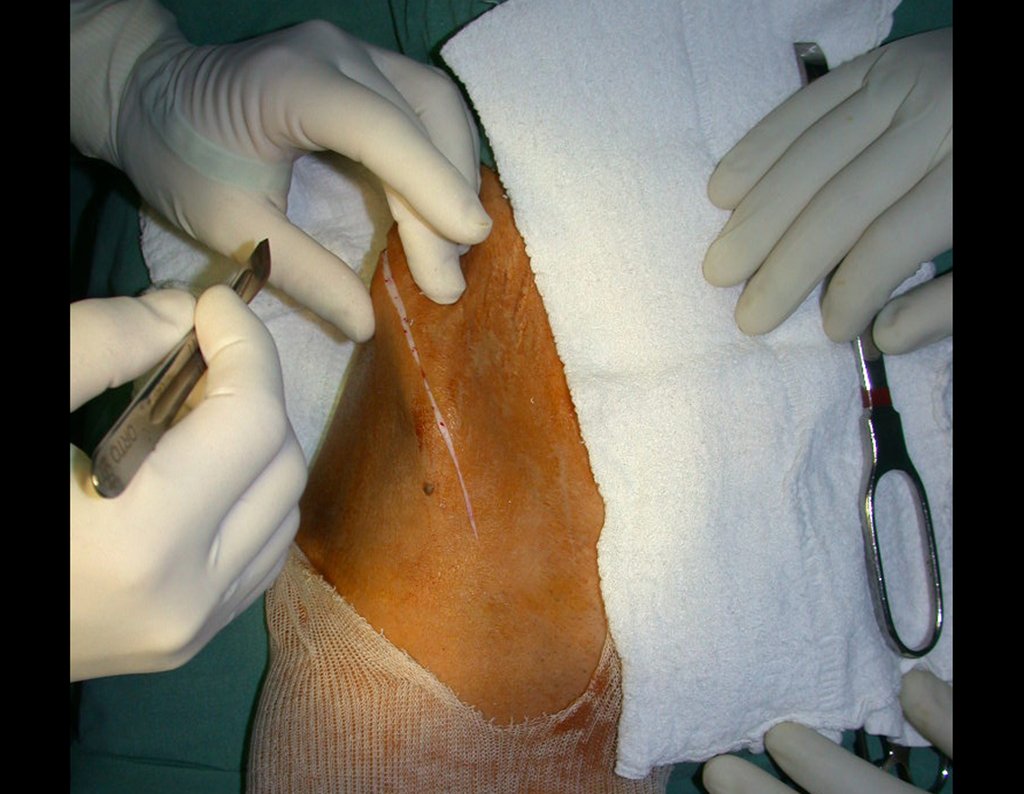
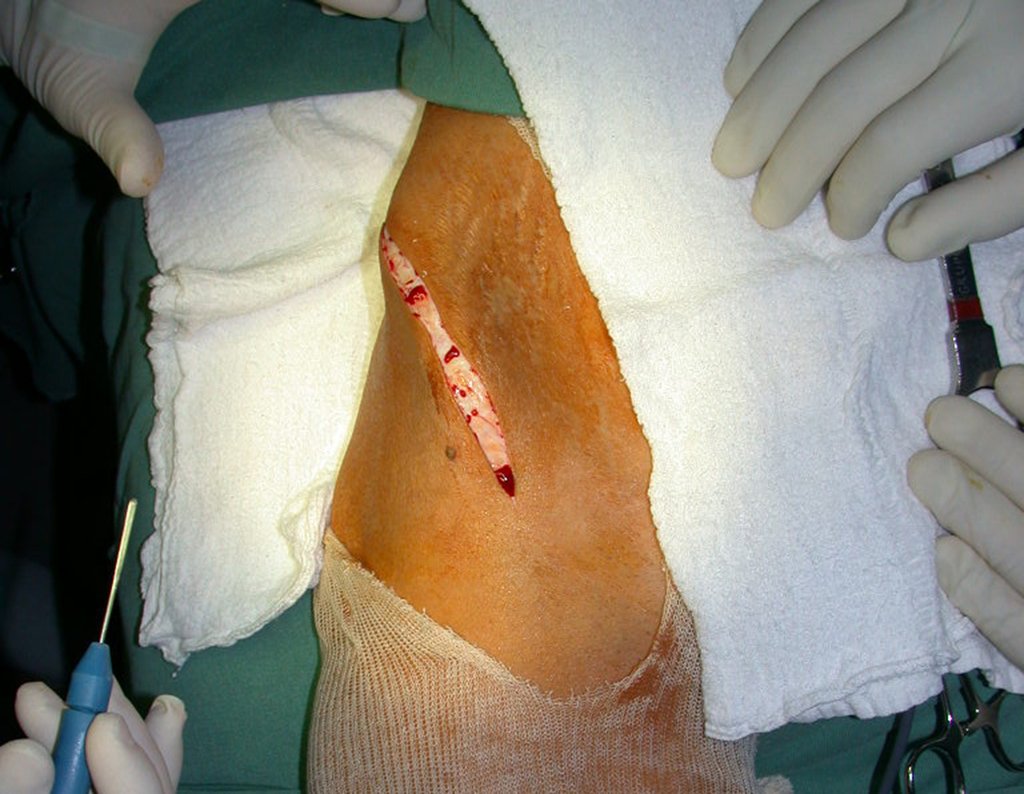
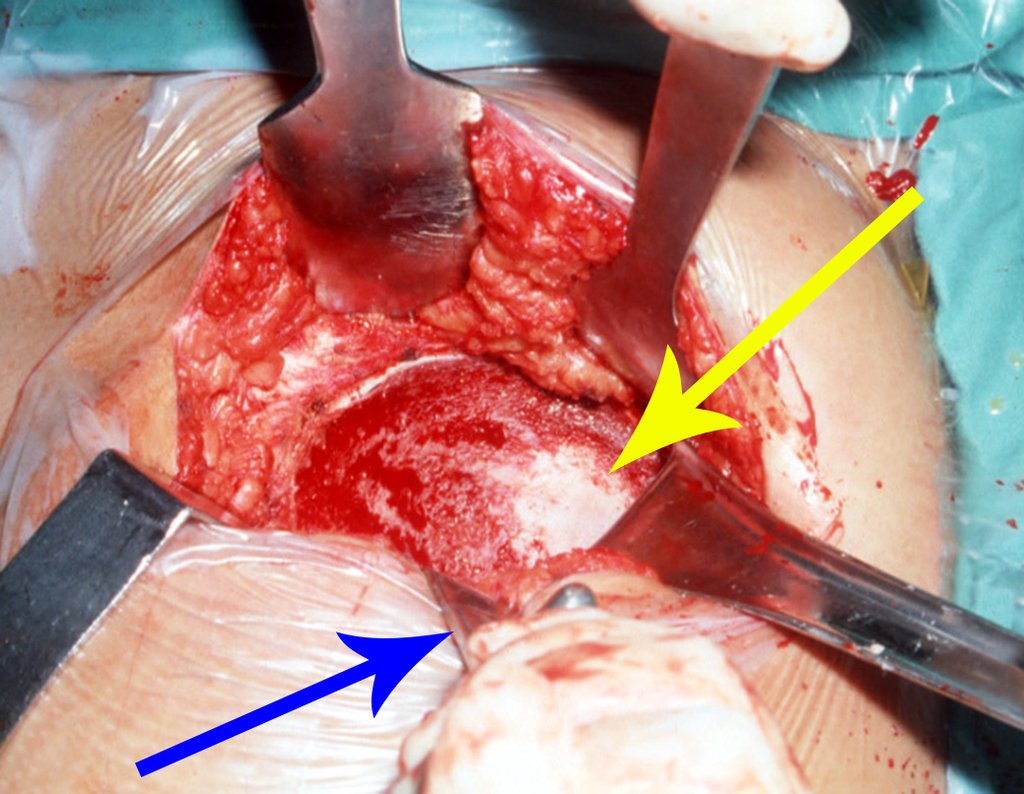
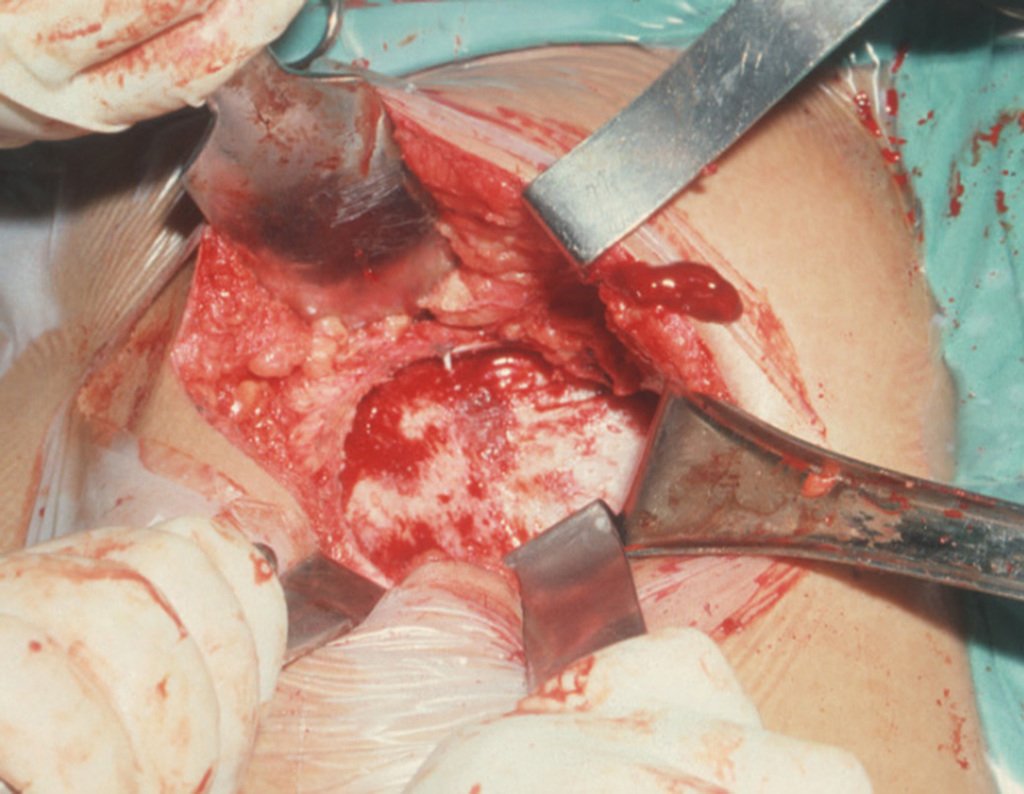
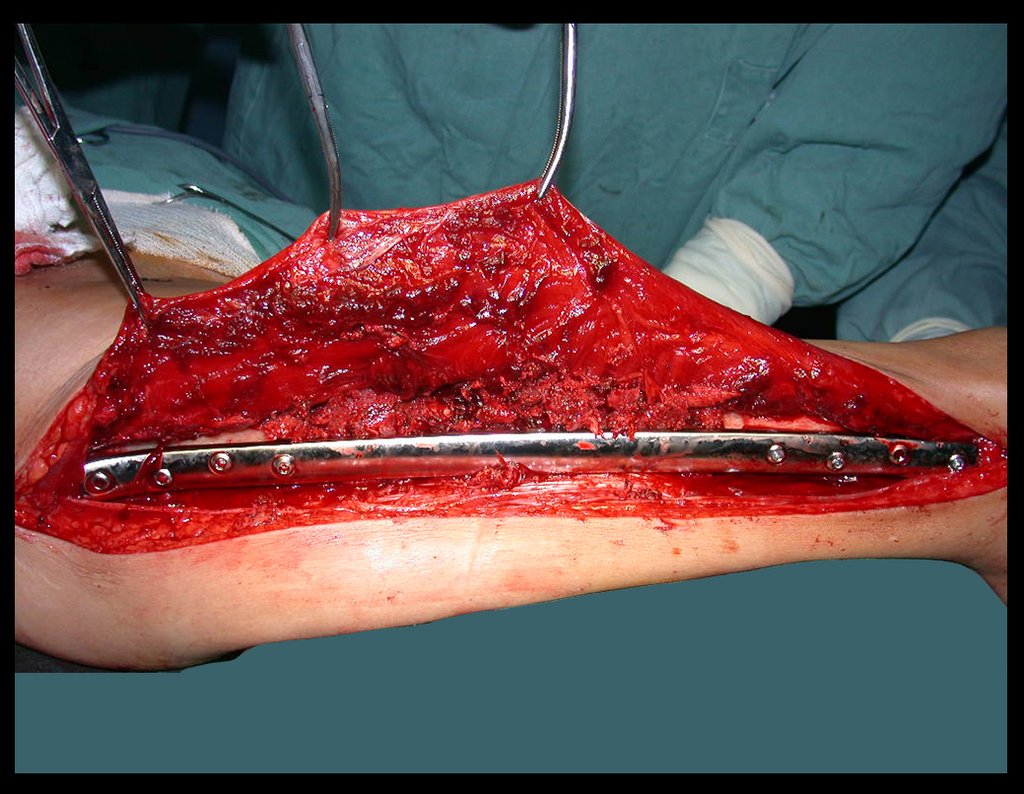
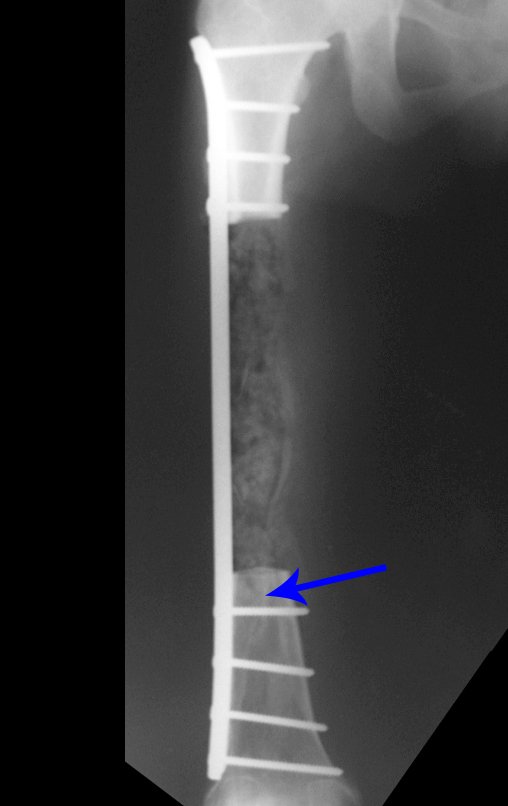
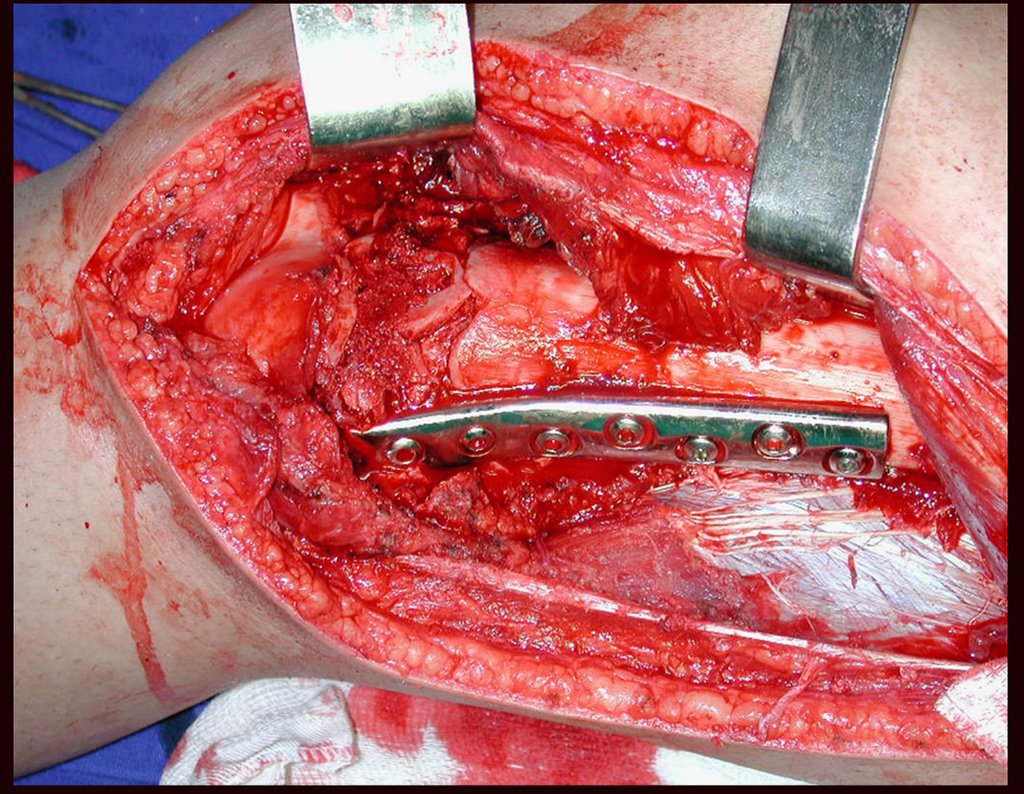
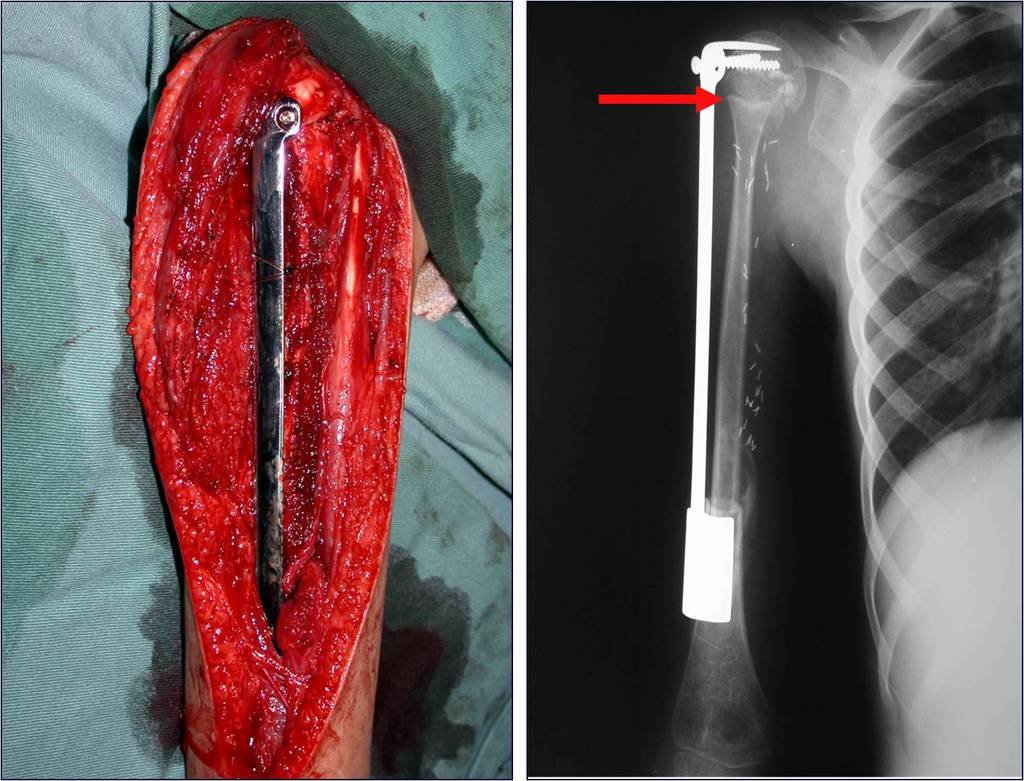
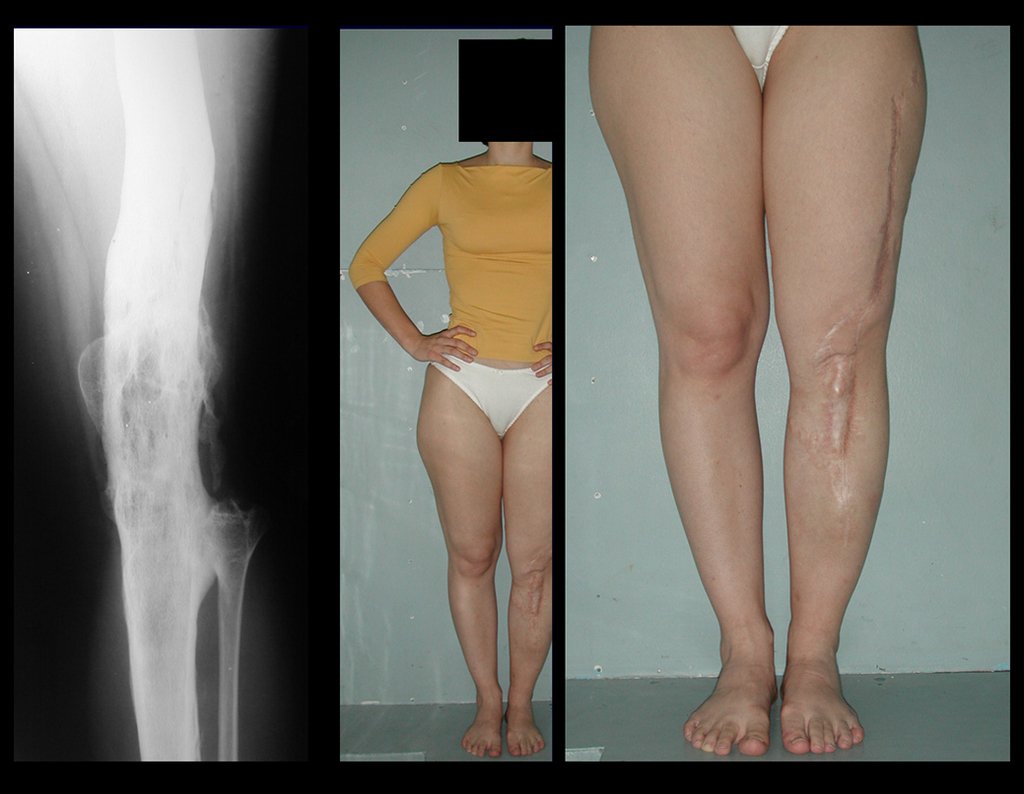
Si en cualquier momento hubiese alteración del cuadro clínico o de la imagen, se debe tratar como condrosarcoma central, realizándose la resección amplia de la lesión y reconstrucción con endoprótesis no convencional, osteosíntesis con injerto autólogo u homólogo o cirugía ablativa conforme a la necesidad de cada caso.
En experiencia de esto autores no es necesario operar un condroma indoloro, cuando es hallazgo casual, sin características de agresividad radiologíca. Realizar curetajeintralesional, con adyuvante local e injerto o cemento, no dará necesidad de observación cuidadosa. En caso de que el examen anatomopatológico de toda el curetaje revele un condrosarcoma, será mucho peor volver a operar esta región ya manipulada quirúrgicamente.
Hay varios casos de ¨condroma¨ que la histología del curetajeintralesional corroboró el aspecto de la biopsia de ¨condroma¨ y sin embargo tuvieron evolución desfavorable. El seguimiento de los exámenes de imagen de estos pacientes revelaran que estaba creciendo ¨nueva¨ lesión local y que se trataba ahora de un condrosarcoma.
En estos curetajes puede ocurrir diseminación local, a distancia y hasta diferenciación a condrosarcoma, empeorando significativamente el pronóstico.
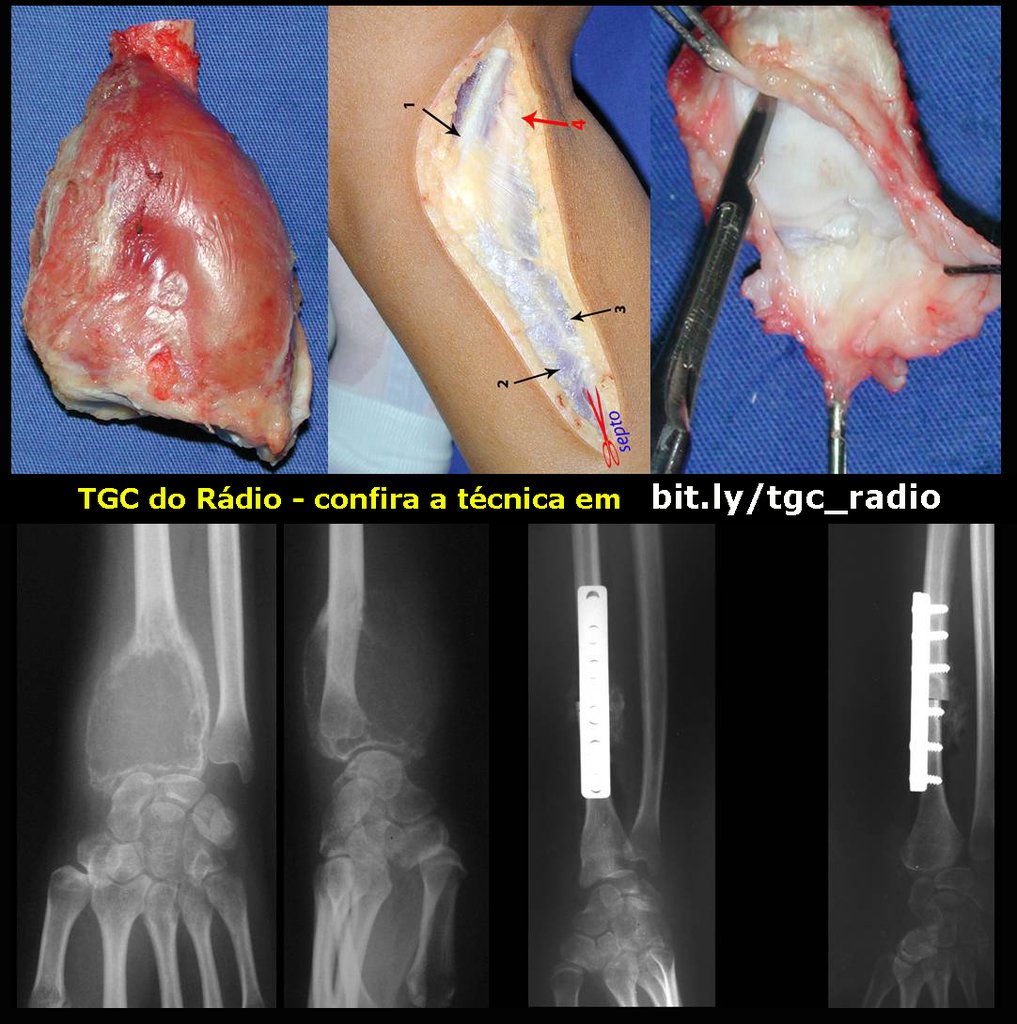
– CondrosarcomaYuxtacortical, el tratamiento es esencialmente quirúrgico, pudiéndose realizar la resección parcial parietal EJEMPLO cuando es posible , procedimiento eficaz y de menor morbilidad en relación a la resección segmentaria.
El perimísio de los tejidos blandos alrededor debe ser removido como margen oncológico, para evitar la recurrencia local.
Es importante resaltar que cuando ocurre crecimiento de una exostosis ósea después de la madurez esquelética, calcificaciones espesas, sin relación con fricción o trauma, probablemente se trate de un condrosarcoma.
Esta situación, una muestra de biopsia negativa no excluye la posibilidad de malignidad de la lesión restante, debiéndose realizar la cirugía de resección con margen oncológico, con especial atención a la superficie de la lesión.
– Condrosarcoma Mesenquimal, tiene la necesidad del control local con cirugía amplia, pudiendo eventualmente tener indicación de quimioterapia con cisplastina y doxirubicina9999.
– Condrosarcoma Desdiferenciado, como el Condrosarcoma de Células claras, se debe realizar el control local con cirugía amplia y quimioterapia con cisplatina y doxirubicina9999.
Complicaciones:
El curetajeintralesional del condrosarcoma puede llevar a la recurrencia local de la enfermedad y a desdiferenciación histológica mas agresiva.
Los casos de condrosarcomadesdiferenciados, las metástasis hematógenas para los pulmones son frecuentes, pudiendo presentar diseminación linfática y recidiva local29. Muchos condrosarcomas presentan tendencia de diseminación local14, consiguiendo tamaños enormes y tornándose inoperables, causando muerte por compresión o complicaciones de la propagación local.
La recidiva local aumenta la incidencia de metástasis pulmomares21.
Bibliografia
1. ACKERMAN, L.V.; SPJUT, H.J. Tumors of bone and cartilage. Atlas of tumor pathology. Washington,Air Force Inst. Pathology, 1962, fasc, 4.
2. CANALE, S.T. Cirurgia ortopédica de Campbell.Barueri: Manole; 2006
3. DAHLIN, D.C. Tumores óseos . Barcelona: Ediciones Toray S/A; 1982
4. DORFMAN, H.D.; CZERNIAK, B. Bone tumors. St Louis, C.V. Mosby Co., 1997, cap. 7, p.410.
5. EDEIKEN, J.; HODES, P.J. Diagnóstico radiológico de las enfermedades de los huesos. Buenos Aires, Panamericana, 1977, cap. 15.
6. ETCHEBEHERE, M. Tumores cartilaginosos malignos: Condrossarcomas. In: Camargo O.P. Clínica Ortopédica. Rio de Janeiro: Med si; 2002. p. 753-759
7. FELDMAN, F. Cartilaginous tumors and cartilage-forming tumor like conditions of the bonés and soft tissues. In: Diseases of the Skeleton System (Roentgen Diagnosis). Part. 6 – Bone Tumors, New York, Springer-Verlag, 1977,p.177.
8. FLETCHER, C.D.M., Unni K.K., OMS – Merters F. (Eds.): World Health Organization. Classification of Tumors. Pathology and Genetics of Tumors of Soft Tissue and Bone. IARC Press: Lyon 2002.
9. GREENSPAN, A. Radiologia ortopédica. Rio de Janeiro: Guanabara; 2001.
10. HENDERSON, E.D.; Le PAGE, G. A. Apud FELDAMAN, F. Cartilaginius tumors and cartilage forming tumor like conditions of the bone and soft tissues. In: Disease of the Skeletal System (Roentgen Diagnosis).
Part. 6 – Bone tumors, New York, Springer Verlag, 1977, p.182.
11. HUVOS, A.G. Bone tumors Diagnosis, Treatment and Prognosis. Philadelphia, W. B. Saunders Co., 1979, p. 13.
12. JAFFE, H.L. Tumores y estados tumorales oseos y articulares. México: La Prensa Medica Mexicana;1966.
13. JESUS-GARCIA, R. – Reynaldo Jesus-Garcia
14. LICHTENSTEIN, L. Barcelona: Talleres Gráficos Ibero-Americanos; 1975.
15. LICHTESTEIN, L. Bone Tumor. 4 Ed St. Louis,C.V. Mosby Co., 1972, cap. 15.
16. LICHTESTEIN, L.; BERNSTEIN, D. Unusual benign and malignant chondroid tumors of bone. Cancer, 12:1142, 1959.
17. MARCOVE, R.C. Condrosarcoma: Diagnóstico y tratamiento. In: Clínicas Ortopécias de Norteamérica. Tumores del aparato musculosquelético. Buenos Aires, Panamericana, 1977, cap. 7.
18. MARCOVE, R.C. et al. Chondrosarcoma of the pélvis and upper end of the femur. Na analisys of factors influencing survival time in113 cases. J. Bone Joint Surg., 54A:61, 1972.
19. MARCOVE, R.C.; SHOJI, H,; HARLEN, M. Altered carbohidrate metabolism in cartilaginous tumors. Contemp. Surg. 5:53, 1974.
20. McFARLAND, G.B.Jr.; McKINLEY, L.M.; REED, R.J. Dedifferentiation of low grade chondrosarcomas. Clin. Orthop., 122:157, 1971.
21. MENENDEZ, L.R. Orthopaedic knowledge update: Actualizaciones en cirugía ortopédica y traumatología. Barcelona: Ars Medica; 2003.
22. O’NEAL, L.W.; ACKERMAN, L. V. Chondrossarcoma of boné. Cancer, 5:551, 1952.
23. PRÓSPERO, J.D. Tumores Ósseos. São Paulo, Roca, 2001, cap. II.
24. ROBBINS. Patologia estrutural e funcional. Rio de Janeiro: Guanabara; 1996.
25. ROMSDAHL, M.; EVANS, H.L.; AYALA, A.G. Surgical treatment of chondrosarcoma. In: Managment of primary bone and soft tissue tumors. Chicago, Year book med. Publisher Inc., 1977, p. 125.
26. ROMSDAHL, M.; Evans, H.L.; Ayala, A.G. Surgical treatment of chondrosarcoma. In: Managment of primary bone and soft tissue tumors. Chicago. Year book med. Publisher Inc., 1977, p.125.
27. SALVADOR, A.H.; BEABOUT, J.W.; DAHLIN, D.C. Mesenchimal chondrosarcoma. Cancer, 28:605, 1971.
28. SCHAJOWICZ, F. Justacortical Chondrosarcoma. J. Bone Joint. Surg., 59B:473, 1977.
29. SCHAJOWICZ, F. Tumores y Lesiones Seudotumorales de Huesos y Articulaciones. Buenos Aires: Editora Médica Panamericana; 1982.
30. TORNBERG, D.N.; RICE, R.W.; JOHNSTON, A.D. The ultrastructure of chondromyxoid fibroma.Clin. Orthop. Rel. Research, 95:295, 1973.
999. J Clin Oncol 30:abstrat 100:23,2012(maluf)
888. Buzaide, A.C.; Maluf, F.C.; Rocha Lima, C.M.
Manual de Oncologia Clinica do Brasil. Dendrix Edição e Design ltda. São Paulo (XI) Sarcomas Ósseos do Adulto, 560-79. 2013